
Living cells don't behave like chemical compounds. Manual operations dominate where automation once reigned. A single contamination event can eliminate a patient's only treatment option. These realities make CGT facility design one of the most demanding undertakings in modern pharmaceutical construction — and they demand that design decisions be made deliberately, early, and with a clear understanding of downstream consequences.
This guide covers the core design considerations that CGT facility stakeholders — developers, manufacturers, process engineers, and their design partners — need to understand before breaking ground.
Key Takeaways
- CGT manufacturing requires fundamentally different facility logic than conventional pharma or biologics production
- Therapy type (autologous vs. allogeneic) determines suite configuration, redundancy requirements, and scale strategy
- Cleanroom classifications, unidirectional flow, and viral vector containment are non-negotiable regulatory design requirements
- Modularity must be designed in from day one — retrofitting is costly and rarely adequate
- A single integrated team spanning architecture, MEP, process, and controls eliminates costly interface gaps
What Makes CGT Facilities Fundamentally Different from Traditional Pharma
The Cell Is the Product
In monoclonal antibody or small-molecule manufacturing, the cell culture is a production tool — a means to an end. In cell therapy, the cell is the final product. That distinction reshapes everything: contamination doesn't just compromise a batch, it eliminates an irreplaceable therapeutic.
This drives an open-processing workflow where operators work directly with product in biosafety cabinets (BSCs) under Grade A conditions, within Grade B background environments. That's a fundamentally different paradigm from the large-scale stainless-steel bioreactors that define conventional biologics plants.
Equipment That Wasn't Designed for This
The CGT equipment landscape is distinctive:
- Biosafety cabinets for aseptic open processing
- Incubator banks for cell expansion and conditioning
- Controlled-rate freezers for cryopreservation
- Single-use bioprocessing systems for closed fluid handling
- Centrifuges and cell washers for intermediate processing steps
Many of these systems were originally developed for blood banking or protein bioprocessing and require design adaptation for GMP CGT applications. ISPE's CGT facility guidance identifies this unique equipment profile as a primary driver of facility differentiation from classical biopharmaceuticals.
Single-use systems in particular require careful design consideration. EU GMP Annex 1 requires risk assessment covering extractables/leachables, fragility, and integrity during cryogenic workflows. These requirements directly shape how single-use systems are specified and integrated into the facility design.
Autologous vs. Allogeneic: Different Facilities for Different Therapies
Therapy type is the single most consequential design input. Getting this wrong means building a facility that works against the manufacturing process from day one.
Autologous: A Logistics and Segregation Problem
Autologous therapies are patient-specific — one manufacturing lot equals one patient. The design implications cascade from that fact:
- Scale-out, not scale-up: Capacity grows by replicating independent suites horizontally, not by increasing batch size
- Chain-of-identity controls: Every suite, every piece of equipment, every sample must be tracked to a specific patient at every step
- Built-in redundancy: Manufacturing disruptions directly affect a named patient, often in an oncology context where time to treatment is measured in weeks
- Suite independence: Cross-contamination between patient lots is a safety event, not just a quality event
ISPE's CGT manufacturing analysis describes an autologous CAR-T example requiring approximately 4,500 m², 3,000 patient batches per year, and 100+ specialized equipment items — compared to an allogeneic example producing 20,000 patient doses per year with substantially less equipment and simpler suite architecture.

Allogeneic: A Capacity and Batch-Scale Problem
Allogeneic therapies use donor-derived or cell-bank-derived starting materials, producing batches that yield multiple patient doses. The facility model looks closer to conventional biologics manufacturing:
- Larger shared processing spaces and bioreactor configurations
- Stronger upstream/downstream capacity planning focus
- More standardized equipment configurations
- Batch release processes that don't require per-patient tracking
Those differences aren't incidental — they define the entire facility logic. Designing an autologous facility using an allogeneic template, or vice versa, produces a facility that fights the manufacturing process rather than supporting it.
Cleanroom Design, Contamination Control & Unidirectional Flow
Classification Requirements
Open cell therapy processing typically requires:
- Grade B background environments (ISO 7 equivalent) for the room
- Grade A zones (ISO 5 equivalent) at biosafety cabinets and isolators where product is exposed
EU GMP Annex 1 defines Grade A as requiring unidirectional airflow at 0.36–0.54 m/s and adjacent Grade B rooms maintaining at least 10 Pa pressure differential. FDA's aseptic guidance maps critical exposed operations to no more than 3,520 particles/m³ ≥ 0.5 μm — the ISO 5 threshold.
These classifications aren't abstract quality metrics. They directly govern:
- HVAC design and air change rates
- Personnel gowning requirements and gowning room placement
- Room pressurization cascades
- Monitoring equipment placement and frequency
Unidirectional Flow: Architecture as Contamination Control
Personnel and materials must move in one direction (from clean to progressively less-clean zones) without backtracking. This principle must be embedded in the architectural layout before a single wall section is drawn.
Pre-designed intermediate return corridors are essential. They maintain unidirectional flow even as operational needs evolve, preventing the improvised workarounds that create contamination pathways.
Annex 1 also requires airflow visualization studies to confirm no adverse airflow migrates from lower to higher grade areas and no contamination ingress occurs over operators at critical work zones.
Viral Vector Containment: A Special Design Challenge
Viral vector manufacturing introduces requirements beyond standard GMP cleanroom rules. Biosafety Level (BSL) classification, determined by an Institutional Biosafety Committee (IBC) risk assessment under NIH Guidelines, drives decisions about:
- Room pressure differentials (potentially negative pressure, reversing standard GMP logic)
- Exhaust air treatment before release
- Airlock configurations and sequenced layouts for multi-product segregation
- Personnel protective measures and decontamination procedures
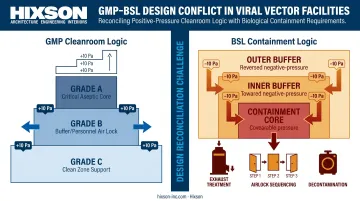
Annex 1 explicitly notes that standard positive-pressure recommendations may need modification for live viral materials, potentially requiring negative pressure with appropriate decontamination before air leaves the production space.
Reconciling BSL containment logic with GMP cleanroom logic is one of the most complex interface problems in CGT facility design. It requires architectural and MEP disciplines to work in parallel from the earliest planning stages — not in sequence.
Designing for Flexibility, Modularity & Scalability
CGT technology is moving fast. Gene editing platforms, closed-system automation, and AI-driven process monitoring are advancing at a pace that makes today's "standard" equipment look dated in five years. Facilities built now must accommodate technologies that haven't been commercially released yet.
Structural Provisions for Future Reconfiguration
Effective flexibility strategies include:
- Standardized building grids that allow room boundaries to shift without structural demolition
- Modular cleanroom wall systems that can be reconfigured as suite configurations change
- Mechanical interstitial levels — a dedicated above-cleanroom maintenance access level where technicians can perform filter changes, valve adjustments, and utility reconfigurations without entering production spaces
An interstitial level eliminates the scenario where routine HEPA filter maintenance forces a production suite shutdown and full requalification. Because production areas are the most utility-intensive spaces in a CGT facility, routing those utilities through an accessible space above the production floor is one of the highest-value design investments available.
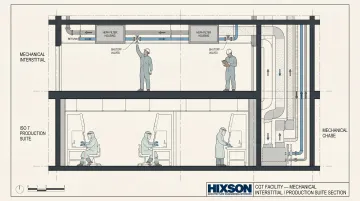
That approach aligns with ISPE's CGT facility guidance, which supports ceiling and wall utility panel systems for power, data, process gases, and liquid waste — designed so equipment layout changes don't require major replumbing or rewiring.
Short-Term Adaptability
For day-to-day operational flexibility:
- Mobile lab benches and height-adjustable countertops for rapid equipment reconfiguration
- Overhead quick-connect utility panels for power, data, and lab gases
- Single-use system integration points that can accept different equipment configurations
Balancing Flexibility with Capital Efficiency
Designing for maximum flexibility and controlling capital cost pull in opposite directions. Phased investment resolves that tradeoff:
- Build the structural provisions now: extra ceiling height, stubbed-in utility connections, oversized mechanical rooms
- Fit out only the capacity you need at opening
- Add capabilities in phases as pipeline and commercial demand justify the spend
Hixson's master planning services for pharmaceutical and biotech clients address phased capital deployment aligned with pipeline progression and regulatory milestones — helping organizations avoid both over-building early and under-building for future scale.
Critical Utilities & Engineering Infrastructure
HVAC: The System That Makes Cleanrooms Work
HVAC isn't a support system in a CGT facility — it's the mechanism by which cleanroom classifications are achieved and maintained. Design priorities include:
- Pressure cascades between classified zones, with 10 Pa differentials as the Annex 1 guidance value
- HEPA and ULPA filtration at appropriate supply and exhaust points
- Unidirectional (laminar) airflow patterns within Grade A zones
- Precise temperature and humidity control — CO₂ incubators and cryogenic equipment place competing demands on room conditions
HVAC system design must be engineered in coordination with room layout, not after it. Trying to retrofit laminar airflow into a room designed around conventional mixed ventilation is expensive and often impossible to validate.
Process Utilities and Electrical Infrastructure
Process utilities for a CGT facility typically include:
- Water for Injection (WFI) and USP purified water systems
- Clean steam for sterilization
- Oil-free compressed air
- Specialty gases: CO₂ for cell culture incubators, nitrogen for inert-atmosphere operations
- Biological waste treatment systems: onsite autoclaves, liquid waste headers, temperature-controlled waste staging for biohazardous single-use consumables
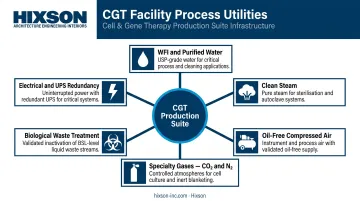
Under-sizing these systems is one of the most common and costly early-stage design errors. Sizing utilities for anticipated future capacity, not just current needs, is far cheaper at design time than adding capacity during operations.
Electrical infrastructure requirements include:
- Redundant power supply and UPS systems for incubators, freezers, and cleanroom pressurization equipment
- Emergency generator capacity with appropriate transfer time specifications
- Arc flash and hazard classification analysis for areas where flammable gases are present
Coordinating HVAC, plumbing, electrical, and controls as a single integrated team — rather than handing disciplines off sequentially — is where most design gaps get caught before they become construction-phase change orders. Hixson's 20 in-house technical disciplines work from shared models and shared timelines, so interface conflicts between mechanical and process engineering surface during design, not after steel is in the ground.
Embedding Regulatory Compliance from the Ground Up
cGMP Cannot Be Retrofitted
FDA 21 CFR Part 211 and 21 CFR Part 600 establish the foundational cGMP framework for drug products and biological products respectively. FDA's human gene therapy CMC guidance adds product-specific requirements for safety, identity, quality, purity, and potency.
Regulators evaluate cleanroom design, documentation, equipment qualification, and process flow as part of product approval. Facilities not designed with cGMP principles from concept stage face:
- Costly remediation before operations can begin
- Re-validation cycles that delay product launch
- Potential rejection of facility submissions
Digital Infrastructure as Regulatory Infrastructure
Electronic batch records (eBR), LIMS, environmental monitoring systems, and process historians are cGMP infrastructure under 21 CFR Part 11 — not software add-ons to be addressed after construction. Designing the physical space to accommodate validated GxP computerized systems from day one means:
- Adequate space and power for server infrastructure
- Data conduit pathways planned alongside utility runs
- Equipment interfaces specified to support audit-trail requirements
Retrofitting validated digital systems into an existing facility typically triggers re-qualification of affected spaces and equipment — extending timelines and adding cost at exactly the point when a facility should be preparing for first-in-human studies.
Frequently Asked Questions
What's the difference between cell therapy and gene therapy?
Cell therapy involves modifying or expanding a patient's cells outside the body before returning them as a therapeutic. Gene therapy introduces genetic material to correct or alter a disease-causing gene — either delivered directly (in vivo) or via modified cells (ex vivo). These distinctions drive substantially different manufacturing processes, equipment requirements, and facility designs.
What are the main challenges in CGT manufacturing?
The primary challenges are highly manual open-processing operations, complex aseptic requirements, patient-specific logistics for autologous therapies, rapidly evolving technology platforms, and maintaining strict chain-of-custody across every production step. CGT facilities must be designed to handle all of these at once.
How are cell and gene therapies manufactured?
Starting materials — patient cells or viral vectors — are processed, expanded, genetically modified where applicable, formulated, and cryopreserved in controlled cleanroom environments under strict cGMP conditions. Autologous and allogeneic workflows differ significantly in suite configuration, batch tracking, and scale strategy.
Is cell and gene therapy growing?
The CGT manufacturing market reached USD 7.28 billion in 2022 and is projected to hit USD 47.1 billion by 2030. The ASGCT Q4 2025 report counted 2,041 gene, cell, and RNA therapies in active development, with 46 FDA-licensed products already on the market.
How much does it cost to manufacture a gene therapy?
Costs vary based on therapy type, batch model, cleanroom grade, automation level, and whether production is in-house or through a CDMO. Key cost drivers include suite footprint, equipment complexity, single-use consumable burden, QC release infrastructure, and biohazardous waste management. Facility design decisions made early in development directly shape these costs.
Is gene therapy legal in the US?
Yes. Gene therapy is legal and regulated in the US under FDA oversight through CBER, with multiple approved products on the market. Manufacturing facilities must comply with cGMP regulations under 21 CFR Parts 211 and 600 and meet product-specific FDA guidance requirements. The regulatory pathway is well-established, with CBER publishing product-specific guidance to support manufacturers through the approval process.


