
Introduction: What Is 21 CFR Part 211 and Why Does It Matter?
Get a cGMP violation wrong and your drug product is legally adulterated — subject to recall, warning letters, and facility shutdown before a single patient complaint is filed. 21 CFR Part 211 is the FDA regulation that defines where that line sits. It establishes minimum Current Good Manufacturing Practice (cGMP) standards for finished pharmaceutical manufacturing and applies to all drug products intended for human or animal use — prescription, OTC, and contract-manufactured alike.
The "current" in cGMP is deliberate. As the FDA notes, manufacturers must use up-to-date technologies and systems; equipment or processes adequate 10–20 years ago fall short by today's standards. Compliance isn't a one-time certification — it's a continuous obligation.
Under 21 CFR §210.1(b), any drug product manufactured, processed, packed, or held in violation of Parts 210, 211, or 212 is considered adulterated under FD&C Act section 501(a)(2)(B). That legal status triggers recalls, warning letters, and facility shutdowns — not just regulatory paperwork.
This guide breaks down who must comply, what each of the 11 subparts actually requires, how facility design and production controls make compliance part of daily operations, and where Part 211 fits relative to Parts 210 and 212.
Key Takeaways
- Part 211 sets minimum cGMP standards for finished pharmaceutical manufacturing — human and animal use — excluding PET drugs and medical gases
- Organized into 11 subparts covering personnel, buildings, equipment, production, labeling, lab controls, and recordkeeping
- Facility design elements — HVAC, plumbing, spatial separation, cleanroom standards — are codified, enforceable requirements
- Non-compliance renders drugs legally adulterated and can trigger Form 483s, warning letters, seizures, or consent decrees
- Works alongside Part 210 (general cGMP framework); Part 212 governs PET drugs separately
Who Must Comply with 21 CFR Part 211?
Regulated Entities
Under §211.1(a), Part 211 applies to the manufacture, processing, packing, or holding of finished drug products for human or animal administration. That scope includes:
- Brand-name and generic drug manufacturers
- Contract manufacturers producing drugs on behalf of other companies
- OTC drug product manufacturers
- Packers and labelers of finished pharmaceuticals
- Holders of finished drug products
FDA's guidance on Quality Agreements for Contract Manufacturing makes clear that quality agreements don't transfer or reduce cGMP responsibility — both the drug owner and the contract facility remain accountable.
Key Exclusions
Three categories fall outside Part 211's scope:
- PET drugs — governed by 21 CFR Part 212
- Medical gases — governed by 21 CFR Part 213
- OTC food/drug combination products — §211.1(c) carves out an exemption for OTC products whose ingredients are ordinarily marketed as food, pending rulemaking proposed in 1978; FDA currently applies Parts 110 and 117 to these products
The Ongoing Compliance Obligation
A facility that met cGMP standards at build-out may not meet them today. Because "current" reflects evolving manufacturing science, manufacturers must continuously review practices as FDA guidance updates. That means facility design decisions — HVAC configurations, process flow layouts, environmental controls — carry compliance implications well beyond initial occupancy. What satisfies an FDA inspection today may require reassessment after the next guidance update.
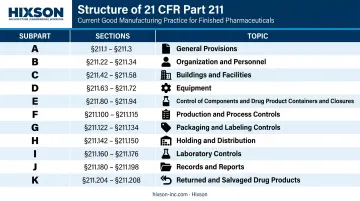
The 11 Subparts of 21 CFR Part 211: A Structured Overview
Part 211 is organized into 11 subparts, each governing a distinct area of pharmaceutical manufacturing control.
| Subpart | Sections | Topic |
|---|---|---|
| A | 211.1–211.3 | General Provisions |
| B | 211.22–211.34 | Organization and Personnel |
| C | 211.42–211.58 | Buildings and Facilities |
| D | 211.63–211.72 | Equipment |
| E | 211.80–211.94 | Components, Containers, and Closures |
| F | 211.100–211.115 | Production and Process Controls |
| G | 211.122–211.137 | Packaging and Labeling Control |
| H | 211.142–211.150 | Holding and Distribution |
| I | 211.160–211.176 | Laboratory Controls |
| J | 211.180–211.198 | Records and Reports |
| K | 211.204–211.208 | Returned and Salvaged Drug Products |
Key requirements by subpart:
- Subpart A establishes scope and references Part 210 definitions. Part 211 supplements — but doesn't supersede — other applicable regulations, including Parts 600–680 for biological products
- Subpart B mandates a dedicated Quality Control Unit (QCU) with authority to approve or reject all components, batch records, and finished products. Personnel must receive ongoing cGMP training; consultants must have documented qualifications
- Subparts C & D govern facilities and equipment — adequate size, spatial separation, validated HVAC, and equipment constructed of non-reactive materials with documented calibration and cleaning
- Subparts E & F trace the control chain from incoming material quarantine through written production procedures, with each step documented and tied to approved specifications
- Subparts G & H extend those controls through tamper-evident packaging, labeling accountability, and FIFO-based distribution with lot-traceable records
- Subparts I–K require scientifically validated lab methods, stability-supported expiration dating, record retention for at least one year post-expiration, and documented procedures for handling complaints, returned products, and salvaged goods
Facility Design, Equipment, and Environmental Controls Under 21 CFR Part 211
Spatial Separation: The Foundation of §211.42
Section 211.42 requires buildings to be of sufficient size, construction, and location to allow cleaning, maintenance, and orderly operations. Process flow — how materials and personnel move through a facility — is a regulatory requirement with direct inspection implications.
Specifically, §211.42(c) mandates physically or procedurally separate, defined areas for:
- Receipt, identification, and pre-release quarantine storage
- Rejected materials awaiting disposition
- Released components, containers, closures, and labeling
- In-process staging and manufacturing
- Packaging and labeling operations
- Pre-release quarantine and post-release product storage
- Laboratory and quality control operations
- Aseptic processing areas
Each zone must be isolated to prevent contamination and mix-ups. The separation requirements are enforceable under FDA inspection, not discretionary design choices.
Aseptic Processing and HVAC Requirements
For aseptic processing areas, §211.42(c)(10) specifies that the facility must include:
- Smooth, hard, cleanable floors, walls, and ceilings
- Temperature and humidity controls
- HEPA-filtered air under positive pressure
- Environmental monitoring systems
- Validated cleaning and disinfection programs
FDA's 2004 aseptic processing guidance adds numeric specificity: higher-cleanliness rooms should maintain at least 10–15 Pa positive pressure relative to adjacent lower-cleanliness rooms with doors closed.
Under §211.46, the HVAC requirements extend further:
- Adequate ventilation controlling air pressure, microorganisms, dust, humidity, and temperature
- Air filtration systems where appropriate
- Separate, completely isolated air handling for penicillin manufacturing — a design requirement with significant facility planning consequences
Plumbing and Computerized Equipment
Section 211.48 requires potable water to be:
- Under continuous positive pressure
- In a system free of defects
- Compliant with EPA Primary Drinking Water Regulations (40 CFR Part 141)
- Connected to drains with adequate sizing and air breaks or equivalent devices to prevent back-siphonage
Section 211.68 governs automated and computerized equipment. Key requirements include:
- Routine calibration under written programs
- Access controls restricting who can modify master production records
- Input/output accuracy checks
- Backup files for all entered data
FDA's 2018 data integrity guidance further defines audit trail expectations under §211.68(b): secure, time-stamped electronic records that capture all changes to critical data.
How Hixson Addresses These Requirements
Designing a Part 211-compliant facility means embedding regulatory decisions — HVAC zoning, plumbing design, cleanroom classification, spatial layout, automation controls — into the project before equipment is purchased or walls are built.
Hixson's team spans architecture, mechanical and electrical engineering, plumbing, process engineering, and controls and automation — all 20 disciplines in-house. The firm engages from the User Requirements Specification (URS) stage through construction administration and commissioning/qualification/validation (CQV).
For aseptic processing, Hixson's scope includes:
- ISO Class 5 critical zone design and unidirectional flow HVAC
- RABS and isolator integration
- Environmental monitoring program design
- Clean utilities: WFI, USP purified water, clean steam, and pure gases
- Alignment with FDA, EU GMP Annex 1, and PIC/S standards

For CDMOs, Hixson has applied this approach on projects such as a nearly 44,000 SF cGMP expansion for Unither Pharmaceuticals' BFS pharmaceutical manufacturing line.
Production Controls, Laboratory Requirements, and Documentation Under 21 CFR Part 211
Production Controls, Laboratory Requirements, and Documentation Under 21 CFR Part 211
Subparts F, G, I, and J of 21 CFR Part 211 govern how drugs are made, labeled, tested, and documented. Together, they form the operational core of cGMP compliance — where facility design decisions directly affect a manufacturer's ability to meet each requirement.
Production and Process Controls (Subpart F)
Sections 211.100–211.115 require that all manufacturing procedures be:
- Written, reviewed, and approved by the QCU before use
- Executed exactly as documented, with any deviation recorded and justified
- Supported by in-process sampling and testing at defined intervals to confirm batch uniformity
Microbiological contamination control procedures must also be written and validated, including all sterilization and aseptic processes.
Once production controls are in place, the focus shifts to how products are packaged and identified before they reach the supply chain.
Packaging and Labeling Controls (Subpart G)
Labeling errors are among the most common causes of FDA warning letters and product recalls. Sections 211.122–211.137 address this directly:
- Labels must be issued, reconciled, and accounted for — unused and obsolete labels destroyed
- Gang-printed labels for different products or strengths are prohibited unless clearly differentiated by size, shape, or color
- OTC human drug products require tamper-evident packaging
- Expiration dates must be supported by stability data
Labeling compliance depends on having reliable data behind every claim — which is where laboratory controls come in.
Laboratory Controls (Subpart I)
Under §§211.160–211.176:
- All lab procedures, specifications, sampling plans, and test methods must be written and followed
- Test methods must be validated for accuracy, sensitivity, specificity, and reproducibility
- Stability testing programs assess product characteristics under defined storage conditions
- Reserve samples must be retained for at least one year after product expiration
Lab data only has value if it's captured, traceable, and reviewable — which brings every requirement back to documentation.
Recordkeeping (Subpart J)
Sections 211.186–211.198 establish the documentation backbone:
- Master Production and Control Records (MPCRs) must be independently checked and signed by two individuals
- Batch Production and Control Records (BPCRs) must document every significant step: equipment used, personnel involved, in-process results, and yield calculations
- Laboratory records must capture all raw data, calculations, and reviewer signatures
- Every batch must pass QCU review before release; any unexplained discrepancy triggers a written investigation that must extend to other potentially affected batches
- Complaint files must be maintained and reviewed for potential safety signals
Taken together, these four subparts make clear that cGMP compliance is not just procedural — it requires physical spaces, systems, and workflows designed to support documentation, contamination control, and traceability at every step.
How 21 CFR Part 211 Relates to Parts 210 and 212
Parts 210 and 211: Read Together
Part 210 establishes the overarching cGMP framework for all drug manufacturing, processing, packing, and holding. Part 211 provides the specific operational requirements for finished pharmaceuticals. The two are inseparable — §210.1(b) makes clear that noncompliance with either Part 210 or Part 211 constitutes adulteration under the FD&C Act.
When other Title 21 regulations overlap (such as Parts 600–680 for biologics or Part 1271 for HCT/Ps), the more specific regulation governs. For combination product manufacturers and biotech companies, this means identifying which regulation controls each aspect of the product before a compliance strategy is built — not after.
Part 211 vs. Part 212: Finished Pharma vs. PET Drugs
| Feature | Part 211 | Part 212 |
|---|---|---|
| Scope | All finished pharmaceuticals (human/animal) | PET drugs only |
| Sterile manufacturing requirements | Yes — full aseptic standards | Not required |
| Radiation shielding | Not specified | Required for PET production |
| Operational flexibility | Structured QCU and batch release | Centralized QC permitted |
Part 212 was designed specifically for Positron Emission Tomography drugs and permits more operational flexibility suited to short half-life production. A facility designed around Part 212 — without aseptic processing rooms, ISO-classified environments, or validated HVAC — cannot simply be repurposed for therapeutic drug production under Part 211 without significant capital investment in infrastructure.
Consequences of Non-Compliance and FDA Enforcement
FDA's enforcement ladder escalates based on severity and persistence of violations:
- Form FDA 483 — Inspectional observations issued after facility inspection, listed in order of risk significance
- Warning Letter — Public advisory action requiring written corrective commitments; serves as prior notice before seizure
- Seizure — Civil action under 21 U.S.C. 334 asking a court to condemn violative products
- Consent Decree / Injunction — Civil judicial process halting violative operations until corrective conditions are met
- Import Alert — Detention without physical examination for facilities with a history of violations
- Criminal Investigation — FDA's Office of Criminal Investigations pursues cases involving intentional FD&C Act violations

The trend is worsening. FDA's FY2024 State of Pharmaceutical Quality report documented 105 warning letters to human drug manufacturing sites for drug quality reasons — up from 94 in FY2023, 72 in FY2022, and 82 in FY2021. FY2024 also saw 75 import-alert additions and 260 recall events involving 421 products.
Common Inspection Triggers
Recurring themes across FDA annual reports and recent warning letters include:
- Failure to investigate out-of-specification results (§211.192) — cited in both the Viatris and Wittman Pharma warning letters (2024)
- Inadequate QCU authority and oversight (§211.22)
- Poor data integrity in laboratory records (§211.68)
- Insufficient cleaning and equipment maintenance records (§211.67)
- Facility design deficiencies that allow cross-contamination risk (§211.42)
A January 2023 DOJ consent decree against LGM Pharma resulted from allegations that adulterated drugs were introduced into interstate commerce under conditions failing to comply with cGMP: the direct statutory consequence of Part 211 violations.
Most of these findings share a common root: facilities that never embedded compliance into their design and operations from the outset. Retrofitting cGMP requirements after a Form 483 observation is significantly more costly (financially and operationally) than building them in from day one.
Frequently Asked Questions
What does 21 CFR Part 211 cover?
21 CFR Part 211 establishes minimum cGMP requirements for finished pharmaceutical manufacturing, spanning personnel qualifications, facility design, equipment standards, production controls, labeling, laboratory testing, recordkeeping, and distribution — for any drug product intended for human or animal use.
What does it mean to be 21 CFR Part 211 compliant?
Compliance means a manufacturer has implemented all required written procedures, quality systems, facility standards, testing protocols, and documentation practices, and can demonstrate this to FDA investigators through records and facility inspections. Compliance is an ongoing operational state — not a certificate you earn once and file away.
What are the key principles of Good Manufacturing Practice?
The five core GMP principles are:
- Quality must be built into a product, not inspected in retroactively
- All critical processes are clearly defined and controlled
- Personnel are qualified and trained for their roles
- Facilities and equipment are maintained and qualified
- Complete records are kept to demonstrate compliance at every step
What are FDA's documentation requirements under 21 CFR Part 211?
All records must be contemporaneous, legible, accurate, and attributable to the individual who performed the activity. Deviations must be recorded and investigated. Batch production records must be retained for at least one year after the product's expiration date.
What is the difference between 21 CFR Parts 210 and 211?
Part 210 is the general cGMP framework applicable to all drug manufacturing operations. Part 211 provides the specific, detailed requirements for finished pharmaceutical products. Both apply simultaneously — noncompliance with either constitutes adulteration under the FD&C Act.
What is the difference between 21 CFR Part 211 and Part 212?
Part 211 applies broadly to finished pharmaceutical manufacturing. Part 212 is specifically designed for PET (positron emission tomography) drugs, which have very short half-lives, and permits operational flexibility not practical under Part 211's broader framework. Manufacturers producing only PET drugs operate under Part 212; those producing other finished pharmaceuticals fall under Part 211.


