
A facility that passes inspection on paper but fails in practice creates risk: contaminated product, patient harm, warning letters, and manufacturing shutdowns. The FDA has made clear that CGMP regulations establish "minimum requirements for the methods, facilities, and controls used in manufacturing, processing, and packing of a drug product" — with the emphasis on minimum.
This guide covers what GMP compliance actually requires in a biotech context, how the core design principles and critical systems work together, how cleanroom classifications translate to operations, and what the design-through-qualification process looks like in practice.
Key Takeaways
- GMP compliance must be designed in from day one — retrofitting it adds cost, time, and revalidation risk
- Unidirectional flow, zone segregation, and surface finish requirements drive every architectural decision
- HVAC, purified water/WFI, clean steam, and environmental monitoring must be fully specified before architecture is finalized
- ISO 5, 7, and 8 map to EU GMP Grades A/B, C, and D — each classification carries distinct operational controls and monitoring obligations
- IQ/OQ/PQ qualification depends heavily on design decisions made months before construction begins
What GMP Compliance Means for Biotech Facilities
The Regulatory Baseline
Good Manufacturing Practice (GMP) — and its "current" variant, cGMP — establishes the minimum requirements for methods, facilities, equipment, and controls needed to ensure a biological product's safety, identity, strength, and purity. The "C" matters: according to the FDA, manufacturers must use technologies and systems that are up-to-date, meaning equipment considered adequate 20 years ago may no longer meet the standard today.
For U.S. biotech manufacturers, three regulatory frameworks govern facility requirements:
- 21 CFR Part 210 — Minimum GMP requirements for drug manufacturing, processing, packing, and holding
- 21 CFR Part 211 — GMP requirements for finished pharmaceuticals, supplementing Parts 600–680 for biologics
- 21 CFR Part 600 — Specific requirements for biological products including vaccines, therapeutic proteins, blood components, and analogous products
International manufacturers or those targeting EU markets must also comply with EU GMP Annex 1 (revised August 2022, effective August 2023), which governs sterile medicinal product manufacture, and ICH Q7, which applies to active pharmaceutical ingredient manufacturing via fermentation and cell culture.
What "Compliant" Actually Looks Like
A GMP-compliant facility is one whose design, construction, systems, and operations can withstand an unannounced regulatory inspection and prevent contamination, mix-ups, and errors at the facility design level.
This contrasts with a standard manufacturing or research lab. The difference shows up in documentation — each of the following must be traceable and defensible:
- Material flow paths and personnel movement routes
- Cleaning cycles and validation records
- HVAC settings and environmental monitoring logs
- Batch records from raw material receipt through final release
A 2025 FDA warning letter to BioStem Life Sciences cited failure to monitor non-viable particulates in ISO 5 areas and failure to validate cleaning in ISO 7 cleanrooms — findings that trace directly back to inadequate facility design and monitoring infrastructure.
Core Design Principles for GMP Biotech Facilities
Unidirectional Flow
GMP facilities require that personnel, materials, and waste follow defined, non-intersecting paths. Cross-traffic creates cross-contamination risk. EU GMP Annex 1 states explicitly that transfer of materials and components into Grade A or B areas must follow a unidirectional process.
In practice, this means:
- Separate corridors for clean and dirty material movement
- Dedicated gowning and de-gowning routes
- Waste removal paths that never intersect product movement
- Airlocks at every zone transition
Flow planning starts at the programming phase. Changing flow paths after construction means tearing out walls and potentially triggering requalification of affected zones.
Zone segregation follows directly from flow planning — once you've defined how people and materials move, you need to define where each operation happens and at what classification level.
Zone Segregation
Biotech facilities are organized around operational zones, each with a defined function, cleanroom classification, and interface requirements:
| Zone | Typical Operations | Classification Range |
|---|---|---|
| Upstream processing | Cell culture, fermentation, bioreactor suites | ISO 8 / Grade D |
| Downstream processing | Chromatography, UF/DF, viral clearance | ISO 7 / Grade C |
| Formulation and fill-finish | Compounding, aseptic filling, lyophilization | ISO 5–7 / Grade A–C |
| Support and ancillary | Buffer prep, equipment staging, gowning | ISO 8 / Grade D |
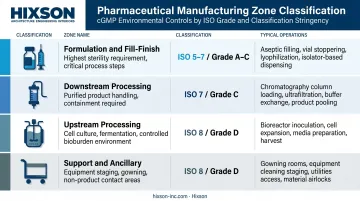
Zone boundaries must be physically enforced through airlocks, pressure differentials, and dedicated access points — not just procedurally defined.
GMP Architectural Requirements
Under 21 CFR 211.42(c)(10), aseptic processing areas require floors, walls, and ceilings that are smooth, hard, and easily cleanable. EU GMP Annex 1 adds that cleanroom surfaces must be "smooth, impervious, unbroken, and easily cleanable to minimize shedding or accumulation of particles or microorganisms."
Key architectural specifications include:
- Coved corners — eliminates right-angle joints where microorganisms accumulate
- Impervious wall and floor finishes — no porous materials, no exposed concrete
- Interlocked airlocks — entry and exit doors cannot open simultaneously; FDA inspection guidance calls for interlocks or audible warning systems
- Pass-through chambers — validated to prevent simultaneous door opening, maintaining pressure differentials between zones
Gowning Room Design
Gowning rooms are where many GMP compliance failures begin. The sequence from street clothes to cleanroom garments must be staged to match the classification level of the cleanroom it serves — a gowning room for an ISO 5 suite requires more staging steps and higher surface quality than one serving an ISO 8 area.
Common regulatory findings tied to gowning design include:
- Inadequate staging space, leading to gown contact with unclean surfaces
- Missing or poorly placed mirrors for self-inspection
- Airlock pressure differentials that don't match the adjacent cleanroom grade
Containment for High-Potency and Biological Hazards
Facilities handling viral vectors, live organisms, or high-potency compounds require containment measures beyond standard GMP architecture. Per CDC/NIH BMBL 6th edition guidance, BSL-3 facilities require:
- Dedicated, ducted mechanical air ventilation with directional airflow from clean areas toward potentially contaminated areas
- Exhaust air that cannot recirculate to other building areas
- Negative pressure suites with independent HVAC exhaust
- Physical separation from adjacent processing zones
For viral vector manufacturing (CAR-T, gene therapy, viral vaccine production), NIH Guidelines require HEPA-equivalent filtration or incineration of exhaust gases from closed systems operating above 10 liters.
Critical Systems Every GMP Biotech Facility Requires
HVAC
HVAC is the backbone of cleanroom compliance. FDA aseptic processing guidance specifies a minimum 10–15 Pascal positive pressure differential between adjacent rooms of differing classification.
Air change rates for ISO 8 spaces require at least 20 changes per hour. ISO 5 zones demand considerably more — EU GMP Annex 1 sets unidirectional airflow velocities at 0.36–0.54 m/s for critical aseptic areas.
Requirements by zone:
- Dedicated air handling units per classification — shared ductwork creates cross-contamination pathways
- HEPA filtration at each supply diffuser in classified spaces
- Validated pressure cascades, typically from highest to lowest classification
- Independent exhaust for containment zones

Purified Water and WFI Systems
Purified Water (PW) and Water for Injection (WFI) are used throughout biotech manufacturing — in buffers, formulation, equipment cleaning, and product contact applications. USP standards set microbial action levels at 100 cfu/mL for PW and 10 cfu/100 mL for WFI.
Design requirements for compliant water systems:
- Recirculating loops maintained at temperature to inhibit biofilm growth
- Elimination of dead legs (stagnant pipe branches where microorganisms accumulate)
- Validated hot water or chemical sanitization cycles
- Validated online conductivity and TOC monitoring per USP standards
Process Utilities
Water systems don't operate in isolation. GMP facilities depend on a full suite of validated process utilities — each with its own purity specifications, qualification requirements, and documentation burden:
- Clean steam — for equipment sterilization via SIP (sterilize-in-place); must meet purity requirements equivalent to WFI when condensed
- Compressed air — oil-free, validated per ISO 8573-1 purity classes for product-contact applications
- Nitrogen — used for tank blanketing and aseptic transfers; must be validated for purity
- CIP/SIP systems — designed with cleanable geometry, validated cycle parameters, and documentation that supports regulatory review
Electrical and Controls Infrastructure
GMP electrical design goes beyond standard commercial requirements. Critical elements include:
- Hygienic or explosion-proof fixture ratings where chemical exposure or flammable materials are present
- UPS and emergency backup power for critical equipment (incubators, cold storage, environmental monitoring systems)
- 21 CFR Part 11 compliance for all electronic records and electronic signatures — requiring audit trails, access controls, and validated system performance for any computer-controlled GMP system
Hixson's electrical engineering team covers hazardous area classification, arc flash studies, and emergency systems design — all relevant capabilities when designing electrical infrastructure for regulated biotech environments.
Environmental Monitoring Infrastructure
Environmental monitoring (EM) infrastructure must be planned into the facility layout from day one — retrofitting sensor networks and sampling ports after construction is costly and often inadequate.
EU GMP Annex 1 Table 6 sets action limits ranging from zero growth in Grade A to 200 cfu/m³ in Grade D for viable airborne contamination, and regulators expect a validated monitoring program tied to those specific limits.
EM infrastructure that must be designed in from the start:
- Viable (microbial) sampling ports at predetermined locations
- Nonviable (particle) monitoring networks with validated sensor placement
- Temperature and humidity sensors with calibrated data loggers
- Pressure differential gauges at every zone transition, with alarming and data capture per 21 CFR Part 11 requirements
Cleanroom Classification: Matching ISO Standards to Biotech Operations
The Classification Framework
ISO 14644-1 defines cleanroom classes by maximum permitted airborne particle counts per cubic meter. For biotech manufacturing, the relevant classes are ISO 5, 7, and 8, which map directly to EU GMP Grades:
| ISO Class | EU GMP Grade | Particles ≥0.5µm (at rest) |
|---|---|---|
| ISO 5 | Grade A / B | 3,520/m³ |
| ISO 7 | Grade C | 352,000/m³ |
| ISO 8 | Grade D | 3,520,000/m³ |
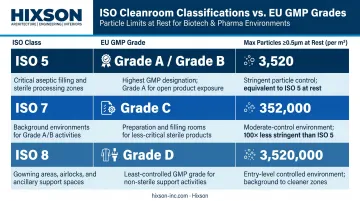
Grade A (ISO 5) is used for the critical zone: the immediate environment around open sterile product, including aseptic filling lines, stopper bowls, and product-contact surfaces. Grade B serves as the background environment surrounding Grade A operations. Grades C and D still require environmental monitoring, but govern progressively lower-risk steps.
Operations by Classification
The appropriate classification for each space depends on the contamination risk to the product — not just the operation's name. In practice:
- ISO 5 / Grade A–B: Aseptic filling, final filtration, open sterile connections, product-contact surface exposure
- ISO 7 / Grade C: Downstream purification, viral clearance, preparation of solutions for aseptic filling
- ISO 8 / Grade D: Upstream processing, buffer preparation, equipment staging, gowning rooms serving classified areas
Validation and Re-Certification
Assigning the right classification is only the starting point. Before a cleanroom can be commissioned, it must complete the full qualification sequence:
- IQ (Installation Qualification) — confirms systems are installed per design specifications
- OQ (Operational Qualification) — verifies systems operate correctly across intended ranges
- PQ (Performance Qualification) — demonstrates consistent performance under actual production conditions
EU GMP Annex 1 requires re-qualification at maximum intervals of 6 months for Grade A and B areas and 12 months for Grade C and D. Re-qualification covers:
- Particle classification testing
- Filter integrity testing
- Airflow verification
- Pressure differential checks
From Blueprint to Qualification: The GMP Facility Design Process
Phased Design and Delivery
GMP compliance requirements must be embedded from the first programming meeting, not layered in during design development. The typical delivery sequence:
- User Requirement Specifications (URS) and Basis of Design — establishes process requirements, classification levels, utility loads, and regulatory framework
- Schematic Design — translates URS into spatial organization, zone layouts, and flow paths
- Design Development — resolves system details, surface specifications, equipment coordination
- Construction Documents — full engineering specifications for construction and procurement
- Construction Administration — oversight to confirm the built facility matches design intent
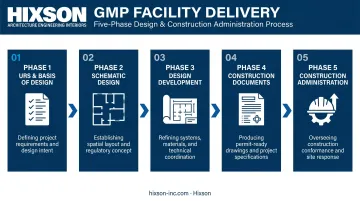
Decisions made in phase one determine what's possible in phases four and five. A gowning room placed incorrectly in schematic design may require a complete layout revision if caught in construction documents — or worse, a costly post-construction retrofit.
The Commissioning and Qualification Sequence
After construction, the qualification sequence begins:
- FAT (Factory Acceptance Testing) — critical equipment is tested at the manufacturer's facility before shipment
- SAT (Site Acceptance Testing) — equipment is re-tested after installation to confirm performance in the actual facility environment
- IQ / OQ / PQ — systems and cleanrooms are formally qualified per the IQ/OQ/PQ sequence
Hixson's Process Engineering and Controls & Automation teams provide FAT and SAT support as part of integrated project delivery, confirming that control systems and process equipment meet performance requirements before and after installation.
Design decisions made months earlier determine how efficiently this sequence completes. Common examples of upstream choices that slow qualification:
- Pipe slopes that prevent complete drainage make SIP validation harder
- Sensors mounted in inaccessible locations complicate calibration
- Surface finishes that trap moisture create EM failures during PQ
The Integration Advantage
When architecture, mechanical, electrical, plumbing, process engineering, and controls operate under one roof, the handoff risk that commonly causes GMP compliance gaps disappears. Siloed teams produce predictable problems:
- Monitoring sensors placed in HVAC dead zones
- Cleanroom diffusers that conflict with equipment layouts
- Electrical penetrations that breach pressure differentials
Hixson's 20 in-house technical disciplines work in parallel from project inception, coordinating GMP requirements across all systems before they require costly rework in the field.
Designing for Scalability and Long-Term Compliance
Why Scalability Is a Regulatory Issue
Adding manufacturing capacity after a facility is validated isn't just a construction project — it's a regulatory event. Under 21 CFR 601.12, biologics manufacturers must report changes to approved applications and assess the effect of any manufacturing change before distributing product made with that change. Capacity additions that affect classified spaces, utilities, or equipment can trigger partial or full requalification.
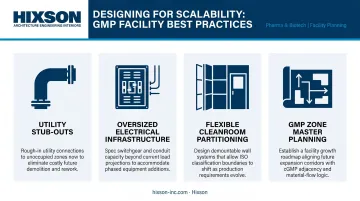
Best practices to minimize that burden at the design stage:
- Utility stub-outs — rough-in connections for future WFI, clean steam, and compressed air drops
- Oversized electrical infrastructure — panels and conduit sized for future load additions
- Flexible cleanroom partitioning — demountable wall systems that can be reconfigured without compromising surface specifications
- GMP zone master planning — formal planning that maps future cleanroom expansion, utility capacity, and phased capital deployment against pipeline milestones before construction begins
Multi-Product and Multi-Modal Facilities
Facilities manufacturing mAbs, cell therapies, and gene therapies — concurrently or sequentially — require additional design attention. Physical segregation between modalities, dedicated versus shared equipment decisions, and campaign changeover procedures all have regulatory implications that must be resolved before the first wall goes up.
Two projects illustrate what this looks like in practice:
- Unither Pharmaceuticals (Rochester, NY) — The initial build accommodated one BFS pharmaceutical manufacturing line, with space and utility capacity allocated for two additional future lines. The second level was built out structurally but held for future lab and office use, preserving optionality without forcing disruptive revalidation when expansion proceeds.
- PETNET Solutions (Siemens Healthineers) — Built for one cyclotron at startup, the 7,500 SF footprint and supporting systems were sized from the outset to accommodate an additional unit — keeping the facility within its validated envelope when that expansion occurs.
In both cases, the decisions that made future scale-up achievable were made during programming, not retrofitted under pressure. Hixson's experience across 75+ years of pharma and biotech facility design has consistently shown that inspection readiness at scale is an outcome of early-stage planning discipline, not late-stage remediation.
Frequently Asked Questions
What does GMP mean in biotech?
GMP stands for Good Manufacturing Practice. The "current" variant (cGMP) requires manufacturers to use up-to-date technologies, systems, and controls — not just follow a fixed standard. In biotech, it represents FDA and international requirements for facilities, equipment, and processes that consistently produce safe, effective biological products.
What does a GMP-compliant facility mean?
A GMP-compliant facility is one whose design, construction, systems, documentation, and operations meet regulatory requirements set by the FDA (21 CFR Parts 210, 211, and 600) and international equivalents like EU GMP Annex 1. It must be inspectable — meaning it can demonstrably and reliably prevent contamination, mix-ups, and errors.
What are the key systems required in a GMP biotech facility?
Regulators expect the following systems to be designed and validated:
- HVAC with HEPA filtration and pressure differential controls
- Purified water and WFI systems
- Clean steam and process utilities (compressed air, nitrogen, CIP/SIP)
- Qualified electrical infrastructure with UPS backup
- Environmental monitoring networks for viable and nonviable contamination
What is the difference between ISO 5, ISO 7, and ISO 8 cleanrooms in biotech?
ISO 5 is the most stringent class, used for open sterile and aseptic operations where product is directly exposed. ISO 7 supports downstream processing and serves as the background environment for ISO 5 zones. ISO 8 covers upstream processing, buffer preparation, and support areas. Each class is defined by maximum allowable airborne particle counts per cubic meter.
How long does it take to design and build a GMP-compliant biotech facility?
Timelines vary widely by project complexity, but a ground-up GMP biotech facility typically requires several years from programming through construction and qualification. Early engagement with an experienced integrated A&E team can compress the design and commissioning phases significantly by reducing coordination delays and catching compliance gaps before construction begins.
What is the IQ/OQ/PQ validation process for a biotech facility?
IQ (Installation Qualification) confirms systems are installed per design specifications. OQ (Operational Qualification) verifies correct operation across the intended range. PQ (Performance Qualification) demonstrates consistent performance under real production conditions. Together, they form the regulatory evidence that the facility is fit for GMP manufacturing.


