
This guide addresses the critical design domains that determine project success: functional zoning, cleanroom classification, pressure regimes, GMP regulatory requirements, building systems, and scalability. Whether you're a project owner, engineer, or architect, this resource provides decision-support for the choices that must be locked before construction begins.
Summary:
- Bulk API facilities require unidirectional flow design, with cleanroom classification (Grade A-D) locked before HVAC design
- Pressure regime (positive for sterile products, negative for hazardous materials) is structurally irreversible once built
- FDA, EU GMP, and WHO regulations converge on 10 Pa minimum pressure differentials and airlock interlocking requirements
- Modular cleanrooms cost $200-400/sq ft vs. $300-600+ for stick-built, with 50-70% labor reduction
- Process validation (Stages 1-3) requires facility qualification before process qualification begins
What Is a Bulk Pharmaceutical Facility?
A bulk pharmaceutical facility manufactures active pharmaceutical ingredients (APIs) — the drug substance at scale, prior to finished dosage form production. ICH Q7 defines an API as "any substance or mixture of substances intended to be used in the manufacture of a drug product and that, when used in production, becomes an active ingredient."
This distinction matters: bulk API manufacturing follows ICH Q7 GMP requirements, while finished dosage facilities (tablets, injectables) fall under separate frameworks like FDA 21 CFR Part 211. The two cannot be conflated — each has distinct cleanroom, containment, and validation requirements.
Product types manufactured in bulk pharma facilities include:
- Sterile APIs (require Grade A/B environments)
- Non-sterile APIs (typically Grade C/D)
- Biologics produced via cell culture or fermentation
- Cytotoxic compounds requiring containment
- Radiopharmaceuticals (negative pressure mandatory)
- Herbal and blood-derived products
Product type determines every downstream design decision: cleanroom classification, pressure regime, HVAC capacity, and segregation strategy. A facility designed for sterile liquid production cannot later manufacture cytotoxic aerosols without complete HVAC redesign and requalification.
The high-level production sequence follows this flow:
- Raw material receipt → quarantine warehouse
- Sampling area (cleanroom with laminar flow hoods)
- Weighing/dispensing for batch preparation
- Production cleanrooms (manufacturing occurs here)
- Secondary packaging and labeling
- Finished goods warehouse → dispatch
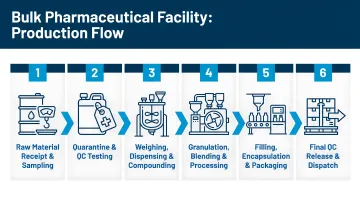
Violations of unidirectional flow are among the leading causes of cross-contamination events — which is why this sequence drives every layout decision that follows.
Core Functional Zones and Flow Design
The principle of unidirectional, linear flow governs pharmaceutical facility layout. Personnel, materials, products, and waste must follow defined, non-intersecting pathways. ICH Q7, Section 4.13 explicitly requires that "the flow of materials and personnel through the building should be designed to prevent mix-ups or contamination."
Personnel flow inward:
- Building entry → primary change room → gowning/airlock → production cleanroom
Product flows outward:
- Production cleanroom → airlock → secondary packaging → finished goods warehouse
Crossing these flows creates contamination ingress points that no amount of cleaning validation can eliminate.
Getting these flows right requires understanding how each functional zone connects to the next. Here's how they sequence in a typical bulk pharmaceutical facility.
Major Functional Zones in Sequence
1. Receiving/Loading Dock Raw materials enter here. Design must include segregated areas for different material types (flammables, narcotics, temperature-sensitive) and quarantine holding pending QC release.
2. Raw Material Warehouse Requires temperature/humidity control with remote alarms, security access controls, and robust wall/door protection for heavy equipment traffic. Pest/vermin exclusion is mandatory.
3. Sampling Area A dedicated cleanroom equipped with laminar flow hoods and material airlocks. This is where incoming QC checks occur before materials enter production.
4. Weighing/Dispensing Area Batch preparation occurs here. This zone must be adjacent to the sampling area and production cleanrooms to minimize material transfer distance.
5. Production Cleanrooms Where API manufacturing occurs. Classification (Grade A-D) depends on product type and must be determined before any other design work begins.
Correct sequencing and physical adjacency of these zones is as important as the spaces themselves. A sampling area positioned far from weighing forces operators to carry materials through uncontrolled corridors — a flow violation that will appear in every FDA inspection report.
Airlock and Gowning Room Design
Airlocks are the pressure transition point between clean corridors and cleanrooms. EU GMP Annex 1 (2022), Section 4.13 mandates interlocked doors that prevent both from opening simultaneously in Grade A/B areas, with visual or audible warnings for Grade C/D.
Critical airlock design requirements:
- Separate personnel airlocks from material airlocks
- Maintain pressure differential consistent with the production room's regime (positive or negative)
- Distinct gown-up rooms (entering cleanroom) from gown-down rooms (exiting)
- Interlocking controls qualified as part of HVAC validation
The airlock pressure must cascade correctly: in positive pressure facilities, cleanroom > airlock > corridor. In negative pressure facilities, the direction reverses.
Secondary Packaging and Finished Goods Warehouse
Secondary packaging applies outer labeling and protective packaging without exposing product to uncontrolled environments. This area requires temperature/humidity control but is typically lower classification than production.
Finished goods warehouse must include:
- Security-controlled access with electronic logging
- Temperature monitoring with remote alarms
- Direct access to outbound loading dock
- Separate secure storage for flammables, narcotics, and radioactive materials per local authority requirements
Modular Facility Design for Multi-Product Manufacturing
Where multiple production cleanrooms are required (multi-product facilities), each cleanroom and its airlock/gowning suite forms a module connected by a clean corridor at neutral pressure. This architecture supports:
- Campaign manufacturing with product changeovers
- Segregation of incompatible products (e.g., penicillin must be separated per 21 CFR 211.42(d))
- Future expansion without disrupting existing operations
Hixson applied this approach at the Unither Pharmaceuticals facility in Rochester, NY: the first level was built to accommodate one Blow, Fill, and Seal (BFS) line immediately, with space pre-allocated for two additional BFS lines in future phases. When Unither was ready to expand capacity, no facility shutdown was required.
Cleanroom Classification and Pressure Regimes
GMP Grade Classification System
The EU GMP Annex 1 (2022) Grade system and corresponding ISO classifications define particle count limits:
| GMP Grade | ISO Class | Application |
|---|---|---|
| Grade A | ISO 5 | Aseptic operations (local zone) |
| Grade B | ISO 7 | Surrounds Grade A |
| Grade C | ISO 8 | Solution prep, filling for terminal sterilization |
| Grade D | ISO 8 | Component washing, assembly |
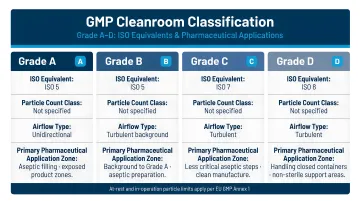
Critical clarification: Bulk API manufacturing does not always require aseptic (Grade A/B) conditions unless the product is sterile. Non-sterile API production may use Grade C or D environments, with classification driven by product sensitivity and contamination risk assessment.
Pressure Differential Requirements
EU GMP Annex 1, Section 4.14 and WHO TRS 1044, Annex 2 both specify a minimum 10 Pa pressure differential between adjacent GMP grades. This ensures air flows from cleaner to less clean areas when doors open.
The pressure cascade architecture:
- Cleanroom → Airlock → Clean corridor → General area
HVAC systems must maintain this cascade at all times. Any lapse shows up immediately in qualification protocols and triggers a non-conformance that halts startup.
Positive Pressure Regime
Used when: Product must be protected from contamination
- Sterile products
- IVs and injectables
- Liquids, creams, dietary supplements
- Herbal medicinal products
- Blood-derived products
Airflow direction: From production room outward
Why it works: Clean air continuously pushes contamination away from the product zone. When doors open, higher-pressure cleanroom air flows into the corridor, preventing ingress of particles or microorganisms.
When the risk equation flips — when the product itself is the hazard — the pressure regime inverts entirely.
Negative Pressure Regime
Used when: Product is hazardous or must be contained
- Aerosols
- Cytotoxic compounds
- Radiopharmaceuticals
- Medicinal gases
- Biologically active compounds at BSL 3/4
Airflow direction: Inward toward production room
Critical requirement: HEPA-filtered exhaust, non-recirculating for the most hazardous compounds
Hixson's design for PETNET Solutions (a 7,500 sq ft facility producing short half-life radioisotopes) required negative pressure containment with specialized exhaust design to meet radiopharmaceutical safety standards.
The regime is permanent. A facility's HVAC system cannot switch between positive and negative pressure post-construction — it is fixed by product type and structurally embedded in ductwork, fan sizing, and exhaust design.
Air Change Rates by Classification
WHO TRS 1010, Annex 8 (2018) provides guidance values:
| ISO Class | Air Changes/Hour |
|---|---|
| ISO 5 | 300-480 |
| ISO 6 | 180 |
| ISO 7 | 60 |
| ISO 8 | 20 |
| Non-sterile areas | 6-20 |
Note: ISO 6 does not map directly to a GMP grade but appears in some specialized pharmaceutical environments. Design for the upper end of these ranges when planning for future flexibility — undersizing HVAC capacity is difficult to correct post-construction.
GMP Compliance and Regulatory Requirements
Primary Regulatory Frameworks
Bulk pharmaceutical facility design is governed by multiple converging frameworks:
United States:
- FDA 21 CFR Part 211, Subpart C mandates buildings of "suitable size, construction and location" with flow "designed to prevent contamination"
- Section 211.42(c)(10) requires aseptic areas to have HEPA-filtered air under positive pressure, smooth/cleanable surfaces, and environmental monitoring
European Union:
- EudraLex Volume 4, Annex 1 (2022) sets particle count limits, pressure differentials, and Contamination Control Strategy (CCS) requirements
Global:
- WHO GMP guidelines (TRS 1044, Annex 2 for sterile products) mirror EU GMP Annex 1 requirements
- ICH Q7 provides the foundational GMP framework for API manufacturing
Biologics/Biosafety:
- CDC/NIH BMBL 6th Edition (2020) governs BSL-3/4 facility design for biologically active compounds
Key insight: While regulations describe what must be achieved, they are often non-prescriptive on how. Design decisions must be justified through Quality Risk Management (QRM) documentation.
Process Validation: A Three-Stage Requirement
Regulatory compliance doesn't stop at facility construction. FDA Process Validation Guidance (2011) embeds facility design directly into the validation lifecycle:
Stage 1 — Process Design: The commercial manufacturing process is defined through URS, P&IDs, and flow diagrams
Stage 2 — Process Qualification: Has two explicit elements:
- Design of the facility and qualification of equipment/utilities (IQ/OQ)
- Process Performance Qualification (PPQ)
"It is essential that activities performed to assure proper facility design and commissioning precede PPQ." — FDA Process Validation Guidance (2011)
Stage 3 — Continued Process Verification: Ongoing assurance is maintained through routine production monitoring
Critical implication: Facility qualification is a regulatory prerequisite to commercial manufacturing. Cleanroom classification, HVAC qualification, and environmental monitoring system placement must be designed in from day one, not retrofitted.
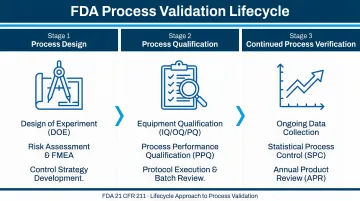
Hixson's integrated team spans 20 in-house technical disciplines — including process engineering, controls/automation, and commissioning support — so validation requirements are identified and resolved during URS and design phases, before they become startup problems.
Quality Risk Management (QRM) in Facility Design
ICH Q9 (2005), Section II.4 specifies QRM applications to facility design:
- Flow of material and personnel
- Minimize contamination
- Open vs. closed equipment
- Cleanrooms vs. isolator technologies
- Dedicated or segregated facilities/equipment
Common QRM tools used in facility design include:
- FMEA (Failure Mode and Effects Analysis)
- HAZOP (Hazard and Operability Study)
- PHA (Preliminary Hazard Analysis)
How this works in practice: Flow diagrams of personnel, materials, products, and waste identify potential cross-contamination ingress points. Design solutions (airlocks, closed systems, pressure differentials, HVAC separation) are selected based on QRM outputs and documented in the CCS.
Note: QC laboratories must be physically separated from production areas with independent air supplies to prevent sample cross-contamination.
Building Systems: HVAC, Finishes, and Infrastructure
HVAC Requirements
WHO TRS 1010, Annex 8 (2018) states HVAC design "should be considered at the initial design stage of a pharmaceutical manufacturing plant" because it "influences architectural building design and layout."
Each classified zone requires:
- Independently designed air handling
- Defined air change rates by GMP grade
- HEPA filtration (minimum)
- Temperature control (default 20°C where not otherwise specified)
- Humidity control (default 60% RH where not otherwise specified)
- Remote-monitored audible/visual alarms for pressure, temperature, and humidity
Critical design rules:
- All systems must be fixed, qualified, and alarmed
- Return air grilles positioned at low levels on walls
- Air supply points located to avoid disrupting laminar flow cabinets
- Qualification sequence: DQ → IQ → OQ → PQ
Undersized HVAC is one of the most expensive mistakes in pharmaceutical facility design. For Abbott Nutrition's $400 million Tipp City, OH expansion, Hixson designed a third aseptic processing line with expanded utilities and HVAC capacity built to accommodate future growth — so the facility never faced the costly retrofits that come with getting this wrong at the start.

Once HVAC zoning is established, surface finishes become the next layer of contamination control — and the requirements are equally unforgiving.
Cleanroom Surface Finish Requirements
EU GMP Annex 1 (2022), Section 4.5 requires "smooth, impervious and unbroken" surfaces "to minimize shedding or accumulation of particles or micro-organisms."
Walls:
- Two-coat high-performance epoxy paint OR fully-welded wall vinyl
- Seamless, gap-free, scrubbable
- No recesses difficult to clean
Ceilings:
- Monolithic construction
- Drop-in tiles prohibited in cleanrooms
Floors:
- Homogeneous vinyl with hot-welded joints OR two-coat epoxy
- Floor-wall and wall-ceiling junctions must have minimum 25mm radius cove for cleanability
Work surfaces:
- Smooth, chemical-resistant stainless steel
- Free-standing (preferably mobile) benches
Lighting:
- Flush-mounted, hermetically sealed fixtures
- Accessible only by Allen key for maintenance
- Avoid natural light entirely in production cleanrooms to preserve product viability and maintain temperature stability
Utilities and Infrastructure
Surface finishes address contamination at the envelope level — but the mechanical and digital systems running through that envelope carry equal compliance weight.
Purified Water: USP <1231> specifies Purified Water "must meet requirements for ionic and organic chemical purity and must be protected from microbial contamination." Water system qualification and requalification must follow validated protocols.
IT/Communications Infrastructure:
- CCTV with unobstructed views of production areas
- Intercom systems between pressurized rooms
- Bar coding in warehouses
- Environmental monitoring data integration
Additional infrastructure requirements for every bulk pharmaceutical facility:
- No drains within production cleanrooms or gowning airlocks — these create direct contamination pathways
- Emergency eyewash and safety shower stations positioned outside the cleanroom but within immediate reach
- All critical production equipment on emergency/uninterruptible power supply
- Electronic access controls, intrusion detection, and CCTV throughout the facility
Designing for Future Scale and Partnering with the Right A&E Firm
Design for Flexibility and Future Expansion
A few upfront design decisions cost relatively little at concept stage but can save millions in retrofit costs later:
- Modular cleanroom configurations
- Adequate ceiling height for HVAC expansion
- Structural load-bearing capacity for future equipment
- Spare utility capacity (electrical, water, compressed air)
A site master plan maps not just the current facility but future phases of development. This governing framework ensures all future capital projects align with long-term operational strategy.
Modular vs. Stick-Built Cleanroom Costs:
| Factor | Modular | Traditional Stick-Built |
|---|---|---|
| Cost per sq ft | $200-$400 | $300-$600+ |
| On-site labor reduction | 50-70% vs. traditional | Baseline |
| Scalability | Relocatable, reconfigurable | Fixed, difficult to modify |
Source: BiocrTech Cost Analysis (2026)
Modular construction can reduce project timelines by 30-50%. When speed to market determines product competitiveness, that compression matters. It also shapes how you staff and structure the design team from day one.
The Value of an Integrated A&E Firm
Engaging an integrated architecture and engineering firm for pharmaceutical facility projects reduces coordination risk, shortens schedules, and ensures critical decisions are resolved collaboratively before construction begins.
Benefits of integration:
- Architecture, structural, mechanical, electrical, process, and controls engineering under one roof
- Critical decisions (cleanroom classification, pressure regime, HVAC design, surface specification) resolved before construction, not through change orders
- Project-lifecycle continuity from URS through startup
Hixson's 20 in-house technical disciplines have served pharma and biotech clients including Abbott Nutrition, Unither Pharmaceuticals, and PETNET Solutions. For Unither, the integrated team prevented costly rework by coordinating hazardous material storage, utility mezzanines, and cleanroom HVAC requirements during preliminary design — decisions that, if missed, would have required structural changes during construction.
Five Decisions That Must Be Made Before Design Begins
Locking these decisions late in design is among the leading causes of pharmaceutical capital project overruns:
- Product type and required cleanroom classification (Grade A-D or ISO 5-8)
- Positive or negative pressure regime (structurally irreversible)
- Single vs. multi-product facility configuration (affects zoning and segregation strategy)
- Greenfield vs. retrofit scope (timelines range from 12-24 months for retrofits to 3-5+ years for greenfield)
- Regulatory jurisdiction and applicable GMP standards (FDA, EU, WHO, or multi-jurisdictional)
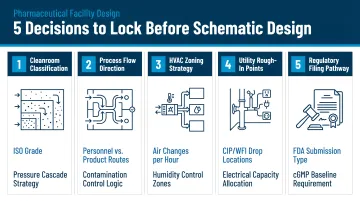
Getting these five decisions locked before schematic design begins is the single most reliable way to protect both schedule and budget on a pharmaceutical capital project.
Frequently Asked Questions
What is the difference between a bulk pharmaceutical facility and a finished dosage facility?
A bulk pharmaceutical facility manufactures the active pharmaceutical ingredient (API) or drug substance at scale, while a finished dosage facility converts that bulk substance into the final patient-ready form (tablets, injectables, etc.). Each has distinct GMP, cleanroom, and containment requirements governed by different regulatory frameworks.
What GMP regulations apply to bulk pharmaceutical manufacturing?
Primary frameworks include FDA 21 CFR Part 211 for US facilities, EudraLex Volume 4 (including Annex 1 for sterile products) for EU compliance, and WHO GMP guidelines globally. The applicable framework depends on target markets and product type.
What cleanroom grade is typically required for bulk API manufacturing?
Requirements vary by product: sterile APIs require Grade A/B controlled environments, while non-sterile bulk APIs may require Grade C or D. The specific classification is driven by the product's sensitivity, manufacturing process, and applicable regulatory guidance.
What is the difference between positive and negative pressure cleanrooms?
Positive pressure rooms push clean air outward to protect the product from contamination (used for sterile products), while negative pressure rooms draw air inward to contain hazardous materials and protect personnel (used for cytotoxics, radiopharmaceuticals, aerosols). The regime is product-specific and cannot be changed after HVAC design is fixed.
How long does it take to design and build a bulk pharmaceutical facility?
Timelines vary by scope: greenfield facilities can take 3–5+ years from concept to startup, while cleanroom retrofits may take 12–24 months. Early-stage decisions on classification, pressure regime, and regulatory framework are the biggest drivers of overall schedule.
What are the most common design mistakes in pharmaceutical facility projects?
The most common errors include:
- Underestimating space needed for HVAC ductwork and air handling units
- Poor zoning that forces operators to cross between clean and non-clean areas
- Locking in cleanroom classification and pressure regime decisions too late in design
- Failing to plan for future capacity or expansion from the start


