
The operational reality is that a poorly designed facility forces compliance to be procedural rather than structural. Staff rely on workarounds. Cleaning validation becomes an ongoing struggle. HVAC systems get retrofitted at significant cost. Regulators notice.
This article examines why facility design is the foundation of GMP compliance — and which specific design decisions carry the most regulatory and operational weight.
Key Takeaways
- GMP compliance is shaped by design decisions: layout, surfaces, and HVAC either support compliance or create constant friction
- The three highest-impact design areas: intentional flow and zoning, cleanable surface and material selection, and integrated HVAC and environmental control
- Skipping GMP design principles leads to regulatory observations, cleaning failures, and costly mid-lifecycle retrofits
- Getting design right from the start costs a fraction of correcting it post-construction
What Is GMP Facility Design?
GMP facility design is the deliberate application of Good Manufacturing Practice principles to the physical layout, materials, systems, and infrastructure of a manufacturing or laboratory facility.
The goal is to make contamination control, cleanability, and environmental monitoring structural features of the building itself — not procedures that depend entirely on staff behavior.
21 CFR Part 211 Subpart C establishes that pharmaceutical manufacturing buildings must be suitable in construction, size, and location, with material and product flow specifically designed to prevent contamination. That same requirement extends, in various forms, across every regulated industry where product safety depends on the physical environment.
Where GMP facility design applies:
- Pharmaceutical and biotech manufacturing (sterile, aseptic, oral solid dose, biologics)
- Food and beverage processing (covered under 21 CFR Part 117)
- Compounding pharmacies (governed by USP General Chapter <797>)
- Medical device manufacturing (21 CFR Part 820 / QMSR)
- Any regulated environment where controlled conditions directly affect product quality and safety
A facility designed for GMP compliance supports consistent product quality and audit readiness on every operating day. The design decisions made before a single wall goes up determine how easy — or how difficult — that becomes.
Key Advantages of GMP-Compliant Facility Design
Each of the following advantages reduces a measurable source of risk, cost, or failure in regulated manufacturing environments. They compound in value over time when applied correctly from the start.
Contamination Prevention Through Intentional Layout and Flow
Contamination pathways in a GMP facility are largely determined by layout. How personnel, materials, and waste move through the space — and whether those routes cross — establishes contamination risk at a structural level that operational procedures alone cannot fully mitigate.
21 CFR 211.42 directly requires that the flow of components, containers, in-process materials, and drug products through buildings be designed to prevent contamination. That's a design requirement, not a procedural one.
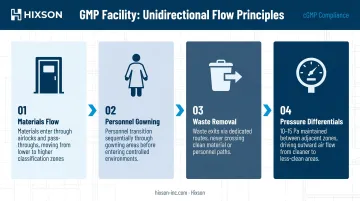
How unidirectional flow works in practice:
- Materials move from lower-classification areas into higher-classification areas only through controlled airlocks or pass-throughs
- Personnel transition through sequential gowning areas before entering classified zones
- Waste exits through dedicated routes that never cross clean material paths
- Pressure differentials — typically 10–15 Pascals between adjacent zones, per FDA aseptic processing guidance and EU GMP Annex 1 — ensure air flows out of cleaner areas, not into them
When this design is absent, cross-traffic creates contamination risks that procedures must compensate for. Those compensating procedures are harder to validate and more difficult to defend during inspections. A 21-year review of FDA enforcement reports found that lack of sterility assurance was the primary driver of sterile product recalls over 2004–2025 — a finding closely tied to contamination control failures.
KPIs impacted: contamination event frequency, deviation report volume, regulatory observations per inspection cycle, batch rejection rate, product recall risk
When it matters most: sterile and aseptic manufacturing, multi-product facilities where different compounds share infrastructure, and food-contact processing environments where a single cross-contamination event can affect an entire product line
Long-Term Cleanability Through Surface and Material Selection
The ability to validate and consistently execute GMP cleaning protocols depends on the surfaces installed during construction. Design decisions made here determine whether cleaning is fast, reliable, and documentable — or slow, inconsistent, and prone to failure.
Regulatory requirements are explicit on this. 21 CFR 211.42 requires floors, walls, and ceilings in aseptic processing areas to have smooth, hard surfaces that are easily cleanable.
USP <797> extends this to sterile compounding buffer areas, requiring surfaces to be smooth, impervious, and free from cracks and crevices — with ceiling-wall junctures coved or caulked and all penetrations effectively sealed.
Design elements that support cleanability:
- Smooth, non-porous wall and floor surfaces (coated concrete, epoxy flooring)
- Coved junctions at wall-to-wall, wall-to-floor, and wall-to-ceiling intersections — eliminating particle-trapping corners
- Flush-mounted lighting and fixtures sealed into ceiling panels
- Corrosion-resistant process equipment with polished, smooth surfaces (per 21 CFR 211.65 requirements for non-reactive, non-absorptive contact surfaces)
- Utility penetrations sealed and minimized within classified spaces

Facilities with inadequate surface design accumulate microbial and particulate contamination in seams, joints, and recesses that standard cleaning protocols cannot adequately reach. Inspectors flag this directly. In a 2018 FDA warning letter, Samson Pharmaceuticals was cited for damaged floors, cracked walls, and peeling paint in an aseptic processing area that was difficult to clean and presented a foreign-matter contamination risk. It was a preventable outcome rooted in surface specification decisions made long before operations began.
Surfaces that can't be adequately cleaned require more labor, more frequent replacement, and generate recurring cleaning validation deviations across the facility's lifecycle.
KPIs impacted: cleaning validation pass rates, cleaning cycle time per room, surface repair frequency, audit findings related to finishes, microbial excursion rates
When it matters most: high-frequency cleaning environments, facilities subject to regular regulatory inspections, and operations where multiple cleaning agents are used — testing both material compatibility and surface durability
Operational Efficiency Through Integrated HVAC and Environmental Control
The HVAC system is the primary engineering control against airborne contamination in a GMP facility. It governs particulate levels, differential pressure between zones, temperature, and humidity. Its effectiveness is almost entirely determined at the design stage, based on how well it integrates with spatial layout and operational requirements.
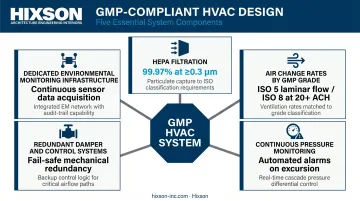
What GMP-compliant HVAC design involves:
- HEPA filtration capable of removing at least 99.97% of particles ≥0.3 µm, per FDA aseptic guidance
- Air change rates calibrated to each zone's GMP grade (Grade A/ISO 5 requiring unidirectional laminar flow; ISO 8 supporting rooms typically requiring at least 20 air changes per hour)
- Continuous pressure monitoring with automated alarms at airlock boundaries
- Redundant damper and control systems to maintain pressure set points through operational fluctuations
- Dedicated environmental monitoring infrastructure built into the design, not retrofitted after occupancy
HVAC systems designed without coordinated input from architecture, process layout, and mechanical engineering result in predictable failures: pressure cascade breakdowns at airlocks, monitoring dead zones where contamination goes undetected, and IQ/OQ/PQ delays that push back commissioning timelines.
The ISPE Baseline Guide Vol. 5 on Commissioning and Qualification frames early integrated C&Q planning as the path to efficient, cost-effective qualification. That's a difficult target when HVAC design is siloed from the broader facility development process.
KPIs impacted: environmental monitoring excursion frequency, HVAC requalification events per year, time-to-qualification for new or renovated spaces, energy consumption per unit of classified area, regulatory observations related to environmental controls
When it matters most: ISO-classified cleanrooms, sterile and aseptic manufacturing, controlled-environment laboratories, and any facility with multiple adjacent GMP grades requiring a defined pressure cascade
What Happens When Facility Design Ignores GMP Requirements
When GMP principles aren't embedded in facility design from the start, the consequences compound. Each remediation cycle costs more than the last, and the regulatory exposure grows with it.
The most common failure patterns:
- Layout and flow gaps create cross-contamination risk that operational procedures must work around. Those workarounds are harder to validate, more prone to human error, and less defensible during inspections
- Surface and material failures prevent cleaning validation from being achieved or sustained. Recurring microbiological excursions, forced cleaning revalidation cycles, and eventual surface replacement are the typical outcomes
- Retrofitted HVAC systems, when designed or modified after construction to meet GMP requirements, frequently produce pressure cascade failures, monitoring gaps, and qualification delays
- Inspector-visible deficiencies generate 483 observations, warning letters, and in serious cases, production shutdowns. In 2024, FDA issued a warning letter to Sun Pharmaceutical following discovery of approximately 450 mL of stagnant liquid in nondedicated manufacturing equipment, with multiple organisms including Pseudomonas stutzeri identified. Multiple batches were recalled. The cited cause was a faulty valve — a design and specification issue, not a random operational lapse

Regulators draw a clear line between design-driven failures and isolated procedural errors. A facility that repeatedly produces contamination events due to structural layout or material specification problems signals a quality system that cannot self-correct — and inspectors treat it accordingly.
How to Get the Most Value from GMP Facility Design
GMP facility design delivers its greatest value when treated as an integrated, multidisciplinary process from the earliest project phases — not as a series of individual decisions made by separate architecture, mechanical, and process teams working in sequence.
The conditions under which GMP design works best:
- Process flow and contamination risk assessment are completed before layouts are finalized
- HVAC, plumbing, and electrical systems are designed in coordination with classified space requirements — not after architectural plans are set
- IQ/OQ/PQ qualification requirements are considered during design, so monitoring infrastructure, access points, and documentation are built in rather than retrofitted
- Engineering disciplines are engaged in parallel from the earliest phases, including definition of the User Requirements Specification (URS)
That integrated approach is how Hixson structures its work. With 20 in-house technical disciplines — architecture, process engineering, mechanical, electrical, plumbing, controls and automation, and construction administration — teams are engaged from project inception in parallel. HVAC design informs architectural layout decisions, and GMP compliance requirements shape every system from the start.
For clients like Abbott Nutrition, Unither Pharmaceuticals, and PETNET Solutions, this coordination has supported cGMP facility work across aseptic processing, sterile fill/finish, and specialized pharmaceutical manufacturing environments.
The same integration that drives good initial design also matters when facilities evolve. GMP compliance isn't a one-time effort — facilities should be reviewed against current standards during renovations, capacity expansions, or product mix changes, because design assumptions made during original construction may no longer hold under new operating conditions.
Conclusion
GMP compliance is an outcome of design. The layout, surfaces, systems, and infrastructure of a facility either make compliance achievable and sustainable, or place it in constant friction with operational realities.
The three design areas with the highest practical impact — intentional flow and zoning, cleanable surfaces and materials, and integrated HVAC and environmental control — each compound in value when applied correctly from the start:
- Reduce audit risk by building compliance into the facility's structure
- Lower operational costs through surfaces and systems that are easier to clean and maintain
- Eliminate expensive mid-lifecycle corrections that arise when design and compliance requirements conflict
For pharmaceutical, biotech, and food and beverage facilities navigating GMP requirements, Hixson brings 75+ years of design and engineering experience across these sectors — helping clients resolve these decisions early, before they become construction problems or audit findings.
Frequently Asked Questions
What is the GMP model in facility management?
GMP in facility management refers to regulatory standards requiring that facilities be designed, operated, and maintained to consistently ensure product quality and safety. This covers layout, environmental controls, cleaning systems, and documentation practices across regulated manufacturing environments.
What is the meaning of GMP design?
GMP design means deliberately engineering a facility's physical infrastructure — including layout, materials, HVAC, and utilities — to meet Good Manufacturing Practice requirements. The goal is to make contamination control, cleanability, and environmental monitoring structural features rather than procedural afterthoughts.
What is a GMP manufacturing facility?
A GMP manufacturing facility is a production environment designed and operated to comply with Good Manufacturing Practice regulations enforced by agencies such as the FDA or EMA. This includes pharmaceutical plants, food and beverage processors, biotech facilities, and compounding pharmacies.
What are the 10 basic principles of cGMP?
The ten core cGMP principles cover quality systems, facility and equipment design, personnel training, process control, documentation, raw material control, production controls, laboratory controls, distribution controls, and change management. Facility design decisions directly shape how well an operation can satisfy most of these requirements from day one.
How does facility layout affect GMP compliance?
Facility layout determines how personnel, materials, and waste move through a space. When those flows are not intentionally designed, they create contamination risks, pressure cascade failures, and procedural workarounds that are difficult to validate and straightforward for regulators to flag during inspections.
What are common GMP facility design mistakes to avoid?
The most frequent mistakes include over- or under-classifying spaces, specifying surface materials that cannot support cleaning validation, failing to integrate HVAC design with classified space requirements, and not aligning facility design with IQ/OQ/PQ qualification requirements from the project's earliest phases.


