
The technical demands are unforgiving. Cleanroom design requires simultaneous mastery of airflow dynamics, pressure cascade logic, surface material selection, and regulatory documentation — all before a single batch is manufactured. Many facilities struggle because these decisions get made in isolation, with mechanical, architectural, and process engineering teams working from different assumptions.
This guide covers the full design framework: ISO 14644 classifications, HVAC and pressure control requirements, facility layout principles, regulatory alignment with FDA cGMP and EU GMP Annex 1, and the validation sequence every GMP cleanroom must complete.
Key Takeaways
- ISO 14644-1 classifies cleanrooms into nine classes (ISO 1–9); pharma applications typically require ISO 5 through ISO 8
- Cleanroom cleanliness rests on three pillars: HEPA filtration, pressure cascades between zones, and non-shedding surfaces
- Pharmaceutical facilities must satisfy ISO 14644, FDA cGMP (21 CFR 210/211), and — for sterile products — EU GMP Annex 1
- Validation — DQ, IQ, OQ, and PQ — is mandatory before production begins
What Is a Pharmaceutical Cleanroom and Why ISO 14644 Matters
ISO 14644-1 defines a cleanroom as a room in which airborne particle concentration is controlled and which is constructed and used to minimize the introduction, generation, and retention of particles, with temperature, humidity, and pressure controlled as necessary.
That definition sounds straightforward. In practice, it represents an entire engineering discipline.
Pharmaceutical cleanrooms operate under stricter controls than semiconductor or aerospace cleanrooms because the failure mode is different. A contaminated drug product — particularly a sterile injectable or biologic — can cause direct patient harm.
That's why regulatory compliance isn't treated as an operational checkbox. It's engineered into every design decision from day one.
ISO 14644's role is to provide a common technical language for particle cleanliness. It gives pharmaceutical companies, regulators (FDA and EMA), and A&E design teams a shared classification framework with defined test methods. When an FDA inspector asks whether an aseptic filling suite meets Grade A requirements, ISO 14644 is the measurement system used to answer that question.
In practical terms, the standard addresses two distinct but related needs:
- Classification: Defining what particle counts are acceptable at each cleanliness level
- Verification: Specifying the test methods used to confirm those levels are being met
ISO 14644 classification alone does not constitute GMP compliance. It establishes how clean the air is. GMP regulations determine what that cleanliness level must be, how it must be monitored, and what operational controls must surround it.
ISO 14644 Classifications and GMP Grade Mapping
The Nine-Class Scale
ISO 14644-1:2015 defines nine cleanroom classes based on maximum allowable airborne particle concentrations per cubic meter. The scale is approximately logarithmic — each class step represents approximately a 10× difference in particle limits at ≥0.5 µm.
| ISO Class | Max particles/m³ ≥0.5 µm | Pharma Application |
|---|---|---|
| ISO 5 | 3,520 | Grade A/B — critical sterile zones |
| ISO 6 | 35,200 | Grade B support / aseptic background |
| ISO 7 | 352,000 | Grade C — sterile background operations |
| ISO 8 | 3,520,000 | Grade D — general pharma manufacturing |

EU GMP Grade Mapping
EU GMP Annex 1 (2022) ties ISO classes directly to its Grade A–D system, but with an important nuance: it specifies particle limits at both rest and in-operation states.
| GMP Grade | At Rest ≥0.5 µm | In Operation ≥0.5 µm | ISO Equivalent |
|---|---|---|---|
| A | 3,520 | 3,520 | ISO 5 |
| B | 3,520 | 352,000 | ISO 5 at rest / ISO 7 operating |
| C | 352,000 | 3,520,000 | ISO 7 at rest / ISO 8 operating |
| D | 3,520,000 | Not predetermined | ISO 8 at rest |
The "at rest" state means HVAC and utilities operating, equipment installed, no personnel present. "In operation" means full production conditions with the maximum number of personnel present. The gap between these two states requires documented justification through risk assessment — regulators expect the rationale, not just the numbers.
Why Both 0.5 µm and 5.0 µm Are Measured
Annex 1 requires monitoring at both particle sizes:
- ≥0.5 µm — broad contamination indicator correlated with microbial risk
- ≥5.0 µm — specifically relevant in Grade A/B because larger particles are more likely to carry viable microorganisms and deposit near open product or containers; Annex 1 notes that elevated 5.0 µm counts can signal early filtration failure or poor practices
Legacy Federal Standard 209E
Federal Standard 209E was formally cancelled on November 29, 2001. Its class nomenclature (Class 100, 10,000, 100,000) still appears in older SOPs and supplier contracts. The FDA's aseptic processing guidance provides the direct equivalences:
- Class 100 → ISO 5
- Class 10,000 → ISO 7
- Class 100,000 → ISO 8
When these legacy terms appear in facility documentation, update them or add explicit cross-references to current ISO class designations — particularly before any regulatory submission or audit readiness review.
Core Engineering Design Requirements: HVAC, Airflow, and Pressure Control
HEPA Filtration and Airflow
HEPA filters — defined by FDA as capturing ≥99.97% of 0.3 µm particles — are the foundation of pharmaceutical cleanroom air quality. But filtration alone isn't enough. How air moves through the space matters as much as how clean it is at the supply point.
Airflow type by grade:
- Grade A (ISO 5): Unidirectional (laminar) flow at 0.36–0.54 m/s at the working position, per Annex 1 — high HEPA ceiling coverage is required
- Grade C/D (ISO 7/8): Non-unidirectional turbulent dilution airflow that flushes the area
Air change rates (ACH) should be calculated based on room volume, occupancy, heat load, and process activity — not pulled from a historical table. FDA guidance indicates ISO 8 support rooms typically require at least 20 ACH, but ISPE supports risk-based air change rate reduction when performance data and qualification support it. That 20 ACH figure shouldn't be applied uniformly to ISO 7 or Grade A/B areas.
Supply and exhaust positioning matters too. Ceiling supply with low-level return at opposing walls creates a "plug flow" effect that maximizes particle removal efficiency.
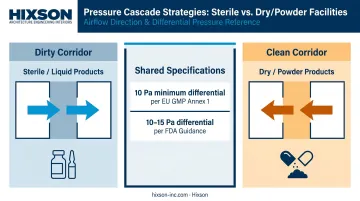
Pressure Cascade Design
Pressure cascades determine how contamination moves between zones when doors open or seals fail. Getting this wrong is one of the most common — and most consequential — cleanroom design errors. FDA 483 observations have cited specific cases where doors wouldn't close properly due to incorrect airflow balance between rooms.
Those failures trace back to the same root cause: cascade philosophy chosen without reference to product type. Two approaches exist:
- Dirty corridor (rooms positive to corridor): Used for sterile/liquid products — keeps organisms out of production rooms
- Clean corridor (rooms negative to corridor): Used for dry/powder products — contains cross-contamination by keeping product dusts inside production rooms
Annex 1 specifies a minimum 10 Pa pressure differential between rooms of different grades. FDA guidance recommends 10–15 Pa between adjacent rooms of differing classification. These are starting points, not universal targets — pressure setpoints and alarm strategies must be defined early in design and verified during qualification.
AHU Design and Temperature/Humidity Control
AHUs serving pharmaceutical cleanrooms typically recirculate approximately 80% of conditioned air through HEPA filtration to maintain temperature and humidity stability. Rooms handling hazardous vapors, potent compounds, or moisture-intensive processes may require 100% single-pass exhaust — which carries significant energy and cost implications.
Temperature and humidity requirements are driven by product stability and process needs, not comfort ranges. Excessive humidity supports microbial growth; very low humidity creates static electricity risks affecting both product and personnel. These parameters must be specified before HVAC design begins — not discovered during commissioning.
All monitoring — pressure differentials, temperature, humidity, and alarm states — must be tied to the Building Management System (BMS) or Environmental Monitoring System (EMS) with documented alarm response procedures.
Pharmaceutical cleanroom HVAC design requires close coordination across mechanical, electrical, plumbing, and architectural disciplines. Fragmented teams commonly introduce pressure-balancing errors and filter housing conflicts that are expensive to fix mid-construction.
Hixson's 20 integrated in-house technical disciplines — covering mechanical, electrical, plumbing, process engineering, architecture, and controls — address this directly. All disciplines work from a single design model rather than reconciling separate deliverables after the fact.
Surface Materials, Facility Layout, and Contamination Zoning
Surface Requirements
Annex 1 is direct: exposed surfaces must be smooth, impervious, and unbroken to minimize particle accumulation and allow effective cleaning and disinfection. The practical implication for design:
- Floors: Epoxy-coated concrete or seamless sheet goods with coved base transitions
- Walls/ceilings: Sandwich panel systems with aluminum or steel facings; no exposed joints or ledges
- Fixtures: Flush-mounted lighting, windows, and doors — no projecting frames or recesses that trap particles
- Doors: Swing toward the higher-pressure room; Annex 1 notes sliding doors may be undesirable because they create recesses that are difficult to clean
Gowning and Transition Zones
Gowning rooms are classification transition zones, not simply places to change clothes. Design must specify garment type (coveralls, gloves, face masks, shoe covers) appropriate to the ISO class being entered. Annex 1 requires airlocks leading to Grade A/B areas to have interlocks preventing entry and exit doors from opening simultaneously. Pass-throughs for materials must be interlocked the same way.
Define the minimum number of personnel in classified areas during the design process — occupancy assumptions directly affect air change rate calculations and particle load modeling.
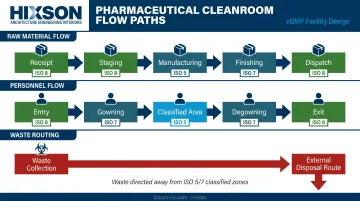
Material and Personnel Flow Zoning
Room adjacency is a contamination control decision, not an architectural preference. The design must trace:
- Raw material receipt → staging → manufacturing → finishing → dispatch
- Personnel entry → gowning → classified area → degowning → exit
- Waste stream routing away from higher-grade zones
Every room adjacency either creates or eliminates a contamination risk. Higher-grade zones should be insulated from lower-grade areas through physical separation and pressure differentials, not just procedural controls. Maintenance access corridors and utility penetrations must be planned to avoid breaching classified environments during routine operations.
Room sequence, door locations, equipment placement, and utility routing are all most cost-effective to resolve during schematic design. After design development, layout changes typically trigger mechanical rework — making early coordination between architecture and engineering disciplines essential.
Regulatory Standards Beyond ISO 14644: FDA cGMP and EU GMP Annex 1
ISO 14644 classifies air cleanliness. It does not govern pharmaceutical manufacturing compliance. Three additional regulatory frameworks apply, often simultaneously:
FDA cGMP (21 CFR Parts 210/211):
- 21 CFR 211.42 requires adequate design and construction, including smooth cleanable surfaces and systems to maintain aseptic conditions
- 21 CFR 211.46 requires adequate control of air pressure, microorganisms, dust, humidity, and temperature, plus appropriate filtration systems
- FDA does not mandate ISO 14644 by name, but inspectors expect facilities to scientifically justify and document classification and monitoring consistently with ISO 14644 or an equivalent methodology
EU GMP Annex 1 (2022, effective 2023):
- The most detailed public-source document for sterile manufacturing requirements
- Adds viable (microbial) particle limits beyond non-viable counts
- Requires Grade A/B environmental monitoring at rest and in operation
- Mandates Grade A/B requalification at least every 6 months and Grade C/D at least every 12 months
- Full compliance is required for EU market access
USP <797>:
- Specifically governs sterile pharmaceutical compounding (not industrial sterile drug manufacturing)
- Specifies ISO 5 primary engineering controls (PEC), ISO 7 buffer rooms, and ISO 8 ante-rooms
Each framework applies to a different scope — USP <797> to compounding operations, FDA cGMP to commercial manufacturing, Annex 1 to sterile products for EU market access. Facilities targeting both US and EU markets must align with FDA cGMP and Annex 1 from the outset; reconciling conflicting requirements after construction is far more costly than resolving them at the design stage.

Designing for dual-market compliance: Hixson's aseptic processing design work explicitly addresses FDA cGMP, EU GMP Annex 1, and PIC/S alignment as a coordinated design requirement — not a post-construction compliance review.
Cleanroom Validation: DQ, IQ, OQ, and PQ
Every GMP pharmaceutical cleanroom must complete a structured qualification sequence before production begins. EU GMP Annex 15 is the clearest public reference for this sequence and the Validation Master Plan (VMP) that governs it.
Design Qualification (DQ)
DQ confirms that the cleanroom design, materials, and engineering systems are documented to meet GMP and process requirements. This phase begins before construction — not after. A DQ that happens post-construction is documentation of what was built, not evidence that what was designed was correct.
Installation Qualification (IQ)
IQ verifies that all systems are installed according to approved design specifications:
- HVAC and AHU installations
- HEPA filter housing and frame integrity
- Monitoring instrument calibration records
- Structural and surface material specifications
Operational Qualification (OQ)
OQ confirms that installed systems perform within their specified operating parameters:
- HEPA filter integrity (DOP/PAO challenge testing)
- Pressure differential verification at all setpoints
- Airflow velocity and uniformity measurements
- Temperature and humidity control range verification
- Air change rate confirmation
- Alarm and interlock functionality
With IQ and OQ complete, the cleanroom has demonstrated it was built correctly and operates as designed. PQ takes the next step: proving it performs under actual use conditions.
Performance Qualification (PQ)
PQ tests the cleanroom under simulated or actual production conditions:
- Particle counts at rest and in operation (both 0.5 µm and 5.0 µm)
- Viable air sampling and surface sampling
- Personnel monitoring during gowning and operation
- Recovery time testing after worst-case disturbances
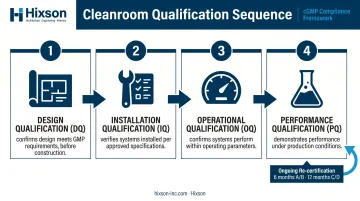
Ongoing re-certification per ISO 14644-2:2015 is required to maintain compliance. Annex 1's requalification intervals (6 months for A/B, 12 months for C/D) set the minimum monitoring cadence.
Hixson engages pharmaceutical and biotech facility projects from URS definition through construction and CQV, with documentation developed to support each qualification phase directly.
Frequently Asked Questions
What is a cleanroom in the pharmaceutical industry?
A pharmaceutical cleanroom is a controlled environment where airborne particle concentrations, temperature, humidity, and pressure are held within defined limits to protect product sterility and quality. These spaces are required for sterile drug manufacturing and are graded based on contamination risk — from highly controlled filling zones to lower-risk support areas.
What are the requirements for a pharmaceutical cleanroom?
Three pillars define pharmaceutical cleanroom requirements:
- Controlled air quality: HEPA filtration, defined air changes per hour, and pressure cascades between grades
- Compliant interior surfaces: smooth, non-porous, chemically resistant, and easy to clean
- Operational controls: gowning protocols, access restrictions, and cleaning SOPs
ISO 14644 classification testing and GMP documentation are required on top of these physical requirements.
How do you design a pharmaceutical cleanroom?
Start with the process: define the manufacturing operation and required ISO/GMP grade. Develop room adjacency and flow diagrams. Engineer the HVAC system, filtration, and pressure cascade. Specify surface materials. Complete DQ documentation before construction begins. Each step informs the next. Starting construction without completed DQ documentation is a direct compliance risk that can force costly redesigns later.
Which regulatory standards apply to pharmaceutical cleanroom design?
ISO 14644 establishes particle cleanliness classification and test methodology. FDA cGMP (21 CFR 210/211) governs facility design and manufacturing requirements in the US. EU GMP Annex 1 applies to sterile product manufacturing for EU market access. USP <797> specifically governs sterile compounding pharmacies.
How do you validate a pharmaceutical cleanroom?
Follow the DQ → IQ → OQ → PQ sequence: DQ confirms the design meets requirements, IQ confirms correct installation, OQ verifies systems operate within parameters, and PQ demonstrates consistent performance under production conditions. Each stage creates a documented audit trail, and ongoing re-certification at defined intervals is required to maintain compliance.
Why are both 0.5 µm and 5.0 µm particle counts required?
The 0.5 µm measurement is a broad indicator of airborne contamination correlated with microbial risk. The 5.0 µm measurement is specifically required in critical (Grade A/B) zones because larger particles are more likely to carry viable microorganisms and deposit near open product or containers. Elevated 5.0 µm counts can also signal early filtration failure before the smaller-size counts show any alarm.


