
Introduction
Pharmaceutical manufacturers face a stark reality: regulatory inspections are won or lost before the inspector walks through the door. A study analyzing 99 GMP inspections across 19 countries uncovered 1,458 deficiencies, with 37% classified as major and 9% as critical. Many of these findings trace back to a single document: the Site Master File. This regulatory document gives authorities their first view of a manufacturer's GMP activities, facility layout, and quality systems.
When inspectors arrive, they use the SMF to conduct their risk assessment and plan which areas to scrutinize. Any discrepancy between what the SMF describes and what they observe on site becomes a major finding.
Yet confusion persists around terminology. Some use "site master plan" and "site master file" interchangeably, while others distinguish between them — and that uncertainty has real consequences for manufacturers preparing documentation.
The regulatory instrument demanded by EU, WHO, and PIC/S authorities is specifically the Site Master File. The decisions made during facility planning directly shape what that file must document.
Summary:
- The Site Master File gives regulatory authorities oversight of GMP activities, facility design, and quality systems at a manufacturing site
- Required by EU/EEA, WHO, PIC/S, TGA, and India's CDSCO—but not mandated by FDA, though still valuable for multi-market manufacturers
- Covers site information, quality management, personnel, premises, equipment, documentation, production, QC, distribution, and self-inspection
- Must be reviewed regularly (industry practice suggests every two years minimum) and updated whenever significant facility or operational changes occur
What Is a Pharma Site Master File — And Why the Terminology Matters
Per PIC/S PE 008-4 and WHO TRS 961 Annex 14, the Site Master File is a factual, comprehensive document prepared by a pharmaceutical manufacturer that describes production and quality control operations at a named site, including any closely integrated operations in adjacent or nearby buildings. It provides specific information about quality management policies, manufacturing activities, and GMP-related operations.
The "site master plan" terminology creates confusion. In pharmaceutical contexts, a site master plan typically refers to the physical layout and infrastructure planning document used during facility design — addressing building footprints, utility routing, expansion zones, and site development. The Site Master File, by contrast, is the regulatory compliance document submitted to authorities.
The two are deeply interconnected. Facility design decisions made during planning directly determine the content and complexity of the eventual SMF. When cleanroom classifications, pressure cascades, and utility systems are designed correctly from the outset, documentation becomes straightforward — because the design decisions already align with regulatory expectations.
International Harmonization
The SMF follows a harmonized format under WHO and PIC/S guidelines, allowing manufacturers to submit the same document to regulatory authorities across multiple markets:
- EU/EEA competent authorities
- Australia's TGA
- India's CDSCO (under revised Schedule M, gazetted January 5, 2024)
- WHO prequalification program
- Any PIC/S member authority
The FDA Exception
The US FDA has no specific CFR provision mandating an SMF. While 21 CFR Parts 210 and 211 establish documentation requirements, they do not reference Site Master Files. However, manufacturers selling into multiple markets should maintain an up-to-date SMF regardless — it gives FDA inspectors a clear operational overview and reduces ambiguity during inspections, even without a formal requirement.
Core Components of a Pharmaceutical Site Master File
WHO guidelines specify that an SMF should not exceed 25-30 pages plus appendices, with schematic layouts and flowcharts preferred over lengthy narratives. This brevity requirement makes every section's accuracy and completeness critical.
The SMF contains nine core sections:
General Information
This section establishes the site's regulatory identity:
- Legal name, physical address, and manufacturing license details
- 24-hour emergency contact number
- GPS coordinates or D-U-N-S identification number
- Brief description of manufacturing activities and product categories
- Applicable manufacturing licenses and regulatory approvals (where not captured above)
This section anchors every subsequent inspection finding to a confirmed, authorized scope.
Organization and Personnel
Inspectors focus heavily on organizational clarity and personnel qualifications:
- Organizational charts for QA, production, and technical operations
- Clearly defined roles, responsibilities, and reporting lines
- Employee training and qualification systems
- Documented qualifications for Authorized Persons and Qualified Persons
- Personnel hygiene and gowning procedures for cleanroom operations
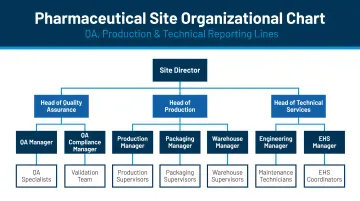
Clear lines of authority are a top inspection focus. Ambiguity here signals systemic quality issues.
Premises and Equipment
This is the most technically complex section, requiring:
- Building layout diagrams with room functional assignments
- Cleanroom classifications (ISO 5-8 or EU GMP Grade A-D) and pressure differentials
- HVAC system descriptions including AHU configurations, air change rates, and filtration levels
- Purified water system details: source, treatment, distribution loop, sanitization method
- Compressed air, clean steam, nitrogen, and other utility systems
- Major manufacturing equipment overview
Regulators expect schematic drawings, not lengthy text. EU GMP Annex 1 (2022) specifies that adjacent rooms of different grades require a minimum pressure differential of 10 Pascals, with continuous monitoring and warning systems. Grade A areas must maintain unidirectional airflow at 0.36-0.54 m/s at working position.
Quality Management System
Modern SMFs must document a comprehensive quality ecosystem:
- Quality policy and objectives
- Self-inspection programs and schedules
- Deviation and CAPA (Corrective and Preventive Action) management
- Document and data control systems
- Change control procedures
- Supplier qualification and approved vendor programs
- Risk management methodology (FMEA, HACCP, or similar)
Data integrity controls per ALCOA+ principles are now a top FDA and EMA inspection focus. ALCOA+ stands for: Attributable, Legible, Contemporaneous, Original, and Accurate — plus Complete, Consistent, Enduring, and Available.
The FDA's December 2018 guidance defines data integrity as "completeness, consistency, and accuracy" and emphasizes audit trails, metadata preservation, and quality culture.
Manufacturing, Quality Control, Distribution, and Self-Inspections
These sections address:
- Main manufacturing processes per product line
- Batch record management and release procedures
- QC testing procedures (physical, chemical, microbiological)
- Distribution controls and recall systems
- Self-inspection programs and follow-up
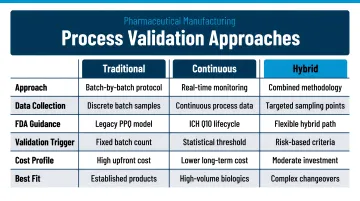
Process validation strategy must be described, referencing one of three approaches per EU GMP Annex 15:
| Validation Approach | Description | Best Suited For |
|---|---|---|
| Traditional | Defined number of batches (typically three consecutive) under routine conditions | Established products with fixed processes |
| Continuous Process Verification | Ongoing monitoring and statistical evaluation of manufacturing performance | Products developed through Quality by Design (QbD) |
| Hybrid | Combines traditional batch confirmation with continuous monitoring | Products where substantial prior knowledge exists |
How Facility Design Shapes SMF Compliance
A pharmaceutical manufacturer's SMF is only as strong as the facility it documents. When facilities are designed with GMP requirements built in from concept through construction documentation, the SMF practically writes itself because design decisions already align with regulatory expectations.
Cleanroom and HVAC Design
The SMF requires documentation of cleanroom classifications, pressure cascades, and air handling specifications. These parameters cannot be retroactively adjusted. Per EU GMP Annex 1 (2022):
| EU GMP Grade | ISO 14644 Equivalent | Max Particles ≥0.5 µm/m³ (at rest) |
|---|---|---|
| Grade A | ISO 5 | 3,520 |
| Grade B | ISO 5 | 3,520 |
| Grade C | ISO 7 | 352,000 |
| Grade D | ISO 8 | 3,520,000 |
Early-stage coordination between architecture, mechanical engineering, and process engineering prevents costly compliance gaps. When these disciplines work in isolation, the resulting facility often requires expensive retrofits to achieve compliance; those retrofits then generate their own documentation burden in the SMF's change control records.
Material, Personnel, and Product Flow
Regulators specifically assess whether facility layout prevents cross-contamination risks. The SMF must demonstrate:
- Logical segregation of production zones
- Dedicated airlocks for personnel and material entry
- Directional flows that prevent backtracking
- Physical separation of incompatible operations
These are architectural decisions that belong in schematic design, not post-construction corrections. Annex 1 specifies separate airlocks for personnel and material, interlocking door systems for Grade A/B areas, and a facility-wide Contamination Control Strategy; it also prohibits sinks and drains in Grade A and B zones entirely.
Utility System Documentation
Purified water, clean steam, compressed air, and nitrogen systems each require qualification data in the SMF. Specifying these systems correctly at the design stage—selecting appropriate materials of construction, loop configurations, and sanitization methods—reduces the validation burden and makes SMF documentation straightforward.
Hixson's integrated structure puts mechanical engineers, architects, and process engineers on the same project team from the start. When the engineer designing the HVAC system and the architect laying out production zones are working from the same set of GMP assumptions, the resulting facility documentation is easier to compile and defensible during inspection.
Renovation and Expansion Scenarios
For manufacturers updating existing facilities, changes to layout, equipment, or utilities must flow through change control and trigger SMF revisions. This is where an A&E partner with direct SMF experience matters most. Renovation projects carry a specific risk: changes that seem straightforward operationally can create undocumented compliance gaps if the design team doesn't understand how each modification maps back to the SMF. An inadvertent gap discovered during an FDA or EMA inspection is far more costly to resolve than one caught at the design review stage.
The SMF's Role in GMP Inspections and Regulatory Compliance
Regulators request the SMF at the outset of a GMP inspection—it is the document inspectors use to conduct their risk assessment and plan which areas to scrutinize. Per PIC/S PE 008-4, WHO TRS 961, and EU GMP Part III, the SMF provides information "useful in general supervision and in the efficient planning and undertaking of GMP inspections."
Any discrepancy between what the SMF states and what inspectors observe on site is a major finding. The most common inspection risk: outdated or inaccurate facility descriptions, especially after renovations or equipment changes.
Which Authorities Require the SMF
| Regulatory Body | SMF Requirement | Authority |
|---|---|---|
| EU/EEA | Mandatory; referenced in EU GMP Chapter 4 | EudraLex Vol. 4, Part III |
| PIC/S | Required; PE 008-4 provides detailed guidance | PIC/S PE 008-4 (2011) |
| WHO | Required for prequalification | WHO TRS 961, Annex 14 |
| Australia TGA | Mandatory for manufacturing license | PIC/S Version 10 adoption |
| India CDSCO | Required under revised Schedule M (2024) | Revised Schedule M |
| US FDA | Not mandated by CFR | 21 CFR Parts 210/211 |
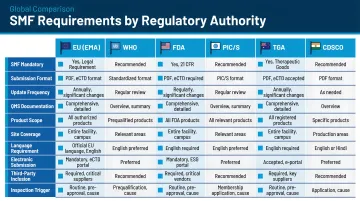
For manufacturers targeting multiple markets, the WHO and PIC/S formats are harmonized — simplifying submissions across jurisdictions without maintaining separate document sets.
Current Regulatory Trends Affecting SMF Content
Data Integrity (ALCOA+): The FDA's December 2018 guidance and EMA's GMP Q&A emphasize that data must be Attributable, Legible, Contemporaneous, Original, and Accurate. Modern SMFs must describe how computerized systems ensure audit trails, metadata preservation, and electronic signature controls.
Lifecycle Approach to Qualification: EU GMP Annex 15 requires a risk-based approach covering the full qualification lifecycle. The SMF must document how DQ, IQ, OQ, and PQ are applied across facilities, equipment, utilities, and processes — not just at initial commissioning.
Computerized System Validation: EU GMP Annex 11 — first effective June 2011 and currently under revision — governs computerized systems across the full lifecycle, including cloud platforms and process control software. The SMF must reference how these systems are validated, maintained, and controlled.
Best Practices for Maintaining and Updating Your SMF
PIC/S PE 008-4, WHO TRS 961 Annex 14, and EU GMP Part III all state the SMF must be "subject to regular review to ensure that it is up to date and representative of current activities." Industry best practice commonly suggests a two-year review interval, though regulatory guidance does not mandate a specific cadence. The requirement is simply "regular review."
Key update triggers:
- Facility modifications (layout changes, cleanroom reclassification, utility upgrades)
- New product introductions or manufacturing process changes
- Equipment installations or replacements
- Organizational changes affecting quality or production leadership
- Regulatory requirement updates
Each annex can carry its own effective date, enabling targeted updates without revising the entire document.
Change Control Integration
All SMF revisions must go through the site's formal change control system, with review and approval by heads of QA, Production, and Technical Operations. Critical procedures include:
- Edition numbering and effective dating for each revision
- Obsolete copy management: stamping, archiving, and removal from circulation
- Controlled distribution list to prevent outdated versions from circulating
- Documentation linking SMF updates to triggering change control records
Three Common SMF Mistakes to Avoid
Even well-maintained SMFs run into recurring problems at inspection. These three issues account for a significant share of SMF-related findings:
- Aspirational descriptions: The SMF must document current operations, not intended future states. Describing systems as they should be—rather than as they are—creates immediate inspection findings.
- Excessive narrative text: Regulators prefer schematic layouts and flow diagrams over dense paragraphs. Simple plans and outline drawings communicate site information faster and with less ambiguity.
- Failure to update after changes: Facility expansions, equipment swaps, or leadership restructuring that don't trigger an SMF update leave a compliance gap between the document and the physical site. This is consistently one of the top sources of inspection deficiencies.

Frequently Asked Questions
What is a site master plan (site master file) in the pharmaceutical industry?
A Site Master File is a regulatory document prepared by a pharmaceutical manufacturer that describes, in factual detail, their GMP-related production and quality control activities at a named site. "Site master plan" is a widely used alternate term; the formal regulatory instrument is called the Site Master File.
What should be included in a site master file?
According to WHO and PIC/S guidelines, a complete SMF covers:
- General site information and quality management system
- Personnel, organization, premises, and equipment
- Documentation systems and production processes
- Quality control, distribution, complaints, and recalls
- Self-inspection programs
What is the validation master plan in GMP?
A Validation Master Plan (VMP) is a site-level document that outlines the overall philosophy, approach, and scope of all validation and qualification activities. The VMP is referenced within the SMF but is a separate document maintained under the quality management system.
What are IQ, OQ, PQ and DQ in pharma?
Under EU GMP Annex 15, each qualification stage has a defined scope:
- DQ (Design Qualification): verifies proposed design suitability
- IQ (Installation Qualification): confirms systems are installed per approved design
- OQ (Operational Qualification): verifies systems perform as intended across operating ranges
- PQ (Performance Qualification): confirms effective, reproducible performance based on approved process methods
What are the 4 types of validation?
The four primary validation types are:
- Process validation: confirms manufacturing processes consistently produce quality product
- Cleaning validation: demonstrates effective removal of residues between production runs
- Computer system validation: ensures software and hardware systems function correctly
- Analytical method validation: proves test methods are suitable for their intended use
What are the 10 golden rules of GMP?
GMP principles generally cover:
- Qualified personnel and validated processes
- Documented procedures and proper facilities/equipment
- Approved raw materials and contamination prevention
- Distribution controls, recall procedures, and self-inspection
- Customer complaint management
The Site Master File is the primary document demonstrating a site's adherence to these principles.


