
Introduction
In pharmaceutical manufacturing, HVAC functions as process control infrastructure — not a comfort system. A poorly designed system can trigger microbial contamination, failed batch releases, FDA warning letters, product recalls, and facility shutdowns.
Unlike standard commercial buildings, pharmaceutical facilities must satisfy overlapping demands simultaneously: strict environmental parameters, cGMP regulatory compliance, contamination control, and comprehensive validation documentation.
None of these requirements can be addressed in isolation. Successful pharmaceutical HVAC design requires close coordination across architecture, process engineering, mechanical engineering, controls, and validation — all of which must align from the earliest design phase.
This guide covers the key parameters that define pharmaceutical HVAC, cleanroom classification and airflow design, system type selection, and the qualification process required before a facility can legally operate under FDA oversight.
Summary:
- Pharmaceutical HVAC controls temperature, humidity, differential pressure, particulate counts, and microbial contamination to cGMP standards—not occupant comfort
- Cleanroom classification (ISO/Federal Standard) determines required air change rates, HEPA filtration levels, and airflow configuration for each manufacturing zone
- System selection (once-through vs. recirculated, constant volume reheat vs. VAV) depends on product type, potency, and contamination risk profile
- Qualification and validation (DQ/IQ/OQ/PQ) are regulatory requirements, not optional commissioning steps
- A pharmaceutical-experienced A/E partner helps avoid costly design conflicts and post-construction modifications
Why Pharmaceutical HVAC Design Is a Different Animal
Standard HVAC systems target human comfort and energy efficiency, governed by standards like ASHRAE 55. Pharmaceutical HVAC, by contrast, targets product safety, process control, and regulatory compliance. The governing standards are cGMP regulations: 21 CFR Parts 210/211 in the U.S., EU GMP Annex 1, and WHO Technical Report Series. Under these frameworks, violations are not minor compliance issues—they can trigger enforcement actions including product recalls, facility shutdowns, and FDA import alerts.
Four Core HVAC Functions Mandated Under cGMP
Pharmaceutical HVAC systems must perform four interdependent functions:
- Filter airborne particulate and microbial contamination via HEPA filtration
- Maintain differential pressure to prevent contaminant migration between zones
- Regulate relative humidity to protect product stability and prevent microbial growth
- Maintain temperature to ensure product integrity and worker safety in heavily gowned environments

Engineering Demands That Set Pharmaceutical HVAC Apart
The scale of the engineering challenge is in a different category entirely. Pharmaceutical cleanrooms require 20–330 air changes per hour compared to 2–10 for standard comfort conditioning. They mandate multi-stage HEPA filtration at 99.97% efficiency down to 0.3 microns, and the building envelope must be designed to be airtight to preserve pressure integrity. According to FDA guidance on aseptic processing, critical sterile operations must be conducted under unidirectional airflow in a Class 100 (ISO 5) environment with airflow velocity of 90 fpm ±20%.
Interdisciplinary Coordination Is Non-Negotiable
Those engineering demands make pharmaceutical HVAC impossible to execute in a single-discipline silo. Each contributor to the facility must be aligned before the design is finalized:
- Process engineers define heat loads and hazardous material handling requirements
- Architects determine room adjacency and airlock layouts
- Controls engineers design Building Automation Systems (BAS) for continuous monitoring and alarm generation
- Validation specialists establish qualification protocols
When that coordination breaks down, the consequences are concrete: pressure zoning conflicts with room adjacency, ductwork clashes with process equipment, and BAS logic disconnects from HVAC design.
Getting the design right the first time is a financial imperative. Post-construction HVAC modifications in a pharmaceutical facility are extraordinarily expensive—involving re-validation, production downtime, and potential regulatory scrutiny. This is where the integrated, in-house structure of a firm like Hixson changes the outcome: with mechanical, process, controls, and architecture disciplines on the same team—not subcontracted—coordination problems surface during design, not during construction.
Critical HVAC Design Parameters for Pharmaceutical Facilities
Temperature and Relative Humidity Control
Pharmaceutical facilities typically maintain temperature ranges of 68–77°F, with a 72°F setpoint common for general manufacturing areas. Heavily gowned personnel in sterile areas may require lower setpoints for operator safety.
Relative humidity (RH) requirements:
- 50±5% RH for most production areas, aligned with ISPE Pharmaceutical Engineering guidelines
- 30±5% RH for spaces handling hygroscopic materials such as certain APIs and oral solid dosage forms
- Below 60% RH to inhibit microbial growth in all controlled areas
RH control must be automatic and continuous—manual adjustment is not acceptable under GMP.
Dehumidification methods:
- Cooling air to dew point using chilled water coils for standard RH control
- Desiccant (chemical) dehumidifiers using silica gel or lithium salt solutions for hygroscopic product areas requiring 30–35% RH
Desiccant systems carry significant space, energy, and maintenance implications — structural loads, regeneration heat demands, and coordination with production scheduling — that must be addressed during early design phases.
Differential Pressure and Pressurization Zoning
Sterile and clean zones must be maintained at positive pressure relative to adjacent less-clean areas, keeping airflow consistently outward and carrying potential contaminants away from product.
EU GMP Annex 1 Section 4.14 mandates a minimum pressure differential of 10 Pa (approximately 0.04 in. water gauge) between adjacent rooms of different grades. Continuous monitoring and prompt alarm systems are required.
Cascading pressure gradient hierarchy:
Main sterile zone → Access corridors → Controlled areas → General areas → Atmosphere
Negative pressure zones:
Hazardous drug (chemo) preparation rooms under USP <800> must be maintained at negative pressure (0.01–0.03 in. w.c.) to all adjacent areas to protect personnel. Negative pressure rooms require 100% exhaust to the outside, significantly increasing make-up air and conditioning demands.
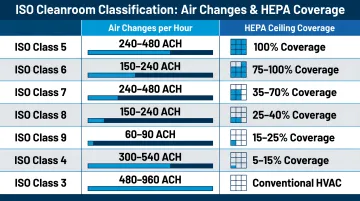
Air Change Rates and HEPA Filtration
The relationship between ISO/Federal Standard class and air change rates (ACH) determines how quickly contaminants are diluted to acceptable concentrations. According to FDA aseptic processing guidance, Class 100,000 (ISO 8) supporting rooms require at least 20 air changes per hour. For cleaner classifications, significantly higher rates are necessary.
Typical air change ranges:
- Class 100,000 (ISO 8): 24–50 ACH at 10–20% HEPA ceiling coverage
- Class 10,000 (ISO 7): 50–100 ACH at 20–40% coverage
- Class 100 (ISO 5): 270–330 ACH at 80–100% HEPA coverage
Multi-stage filtration:
The standard pharmaceutical filtration regime includes four sequential stages:
- Pre-filter (~10 micron/EU4)
- Intermediate filter (~3 micron/EU8)
- Central final filter (~0.3 micron/EU12/HEPA)
- Terminal HEPA filters at supply outlets
HEPA filters rated H13 per EN 1822 achieve ≥99.95% integral efficiency at the most penetrating particle size (MPPS), per AAF pharmaceutical filtration guidance.
Cleanroom Classification, Airflow Patterns, and Pressure Zoning
Cleanroom Classification Systems
Federal Standard 209E and ISO 14644-1 equivalents:
- Class 100 = ISO 5 (critical sterile operations)
- Class 1,000 = ISO 6
- Class 10,000 = ISO 7
- Class 100,000 = ISO 8
Typical pharmaceutical process assignments:
- Sterile injectable manufacturing: Class 100 (ISO 5) within a Class 10,000 (ISO 7) background
- Topical and oral liquid manufacturing: Class 10,000 (ISO 7)
- Tablet and capsule manufacturing: Class 100,000 (ISO 8)
Facilities operating under both U.S. and international standards should note that EU GMP uses a Grade A–D classification system and is generally more stringent than FDA requirements for sterile areas. The 2022 revision of Annex 1 removed the 5.0 µm particle channel for classifying Grade A and Grade B areas at rest — though continuous monitoring of ≥5.0 µm particles remains required for its diagnostic value.
Unidirectional vs. Non-Unidirectional Airflow
Non-unidirectional (turbulent) airflow:
- Used in Class 1,000–100,000 spaces
- Relies on dilution to reduce airborne contamination
- Air supplied through ceiling diffusers with upstream or integral HEPA filtration
- Best suited for spaces where the primary contamination source is external rather than internal
Unidirectional (laminar) airflow:
- Mandatory for Class 100 (ISO 5) critical areas
- Single-pass parallel air streams at 90 fpm ±20% (70–110 fpm) per FDA aseptic guidance
- Contamination generated in the room is swept directly to returns
- Vertical downflow (HEPA ceiling, floor-level or wall-level returns) is preferred over horizontal flow — better suited to pharmaceutical layouts and more protective of operators and product
- At Class 100, 80–100% of the ceiling must consist of HEPA filter units
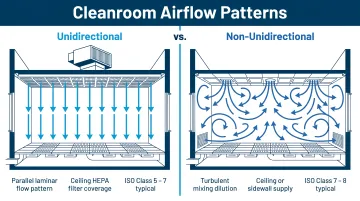
Airflow pattern decisions directly shape how the facility must be laid out — particularly where airlocks are placed and how return air systems are configured.
Building Layout, Airlock Design, and Return Air Placement
Critical layout principles:
- Rooms of similar classification should be physically adjacent to share air handling systems and minimize duct runs
- Pressure gradient must flow unidirectionally from cleanest to least clean zones
- Airlocks between pressure zones are mandatory—no area should open directly into a sterile room
Sterile zone sub-zone hierarchy:
Main sterile zone (white) → Gowning zone (grey/white) → Controlled area → General production
Return air system design:
- Return grilles located low on walls near the floor to capture settling particles
- Symmetrical placement prevents dead air pockets
- Avoid placing grilles near door openings to lower-pressure areas — the pressure differential can draw contaminated air in during door operation
- Manual dampers on return air risers should be operable from non-sterile areas
Pharmaceutical HVAC System Types and Equipment Selection
Once-Through vs. Recirculated Air Systems
Once-through systems:
- Condition outdoor air, supply it to the space, and exhaust it—no recirculation
- Required for: hazardous/potent APIs, bulk pharmaceutical chemical plants, and any area where cross-contamination between products is unacceptable
- Advantages: maximum fresh air supply and no return duct cross-contamination risk
- Disadvantages: very high operating energy costs and heavy filter loading
Recirculated systems:
- Recondition return air within the space or AHU
- Preferred for sterile manufacturing and finished oral solid dosage spaces
- Reduce heating/cooling costs and filter loading while enabling tighter environmental control
Constant Volume Reheat vs. Variable Air Volume
Constant Volume Reheat (CVRH):
- The standard system for classified pharmaceutical manufacturing areas
- Supply air volume remains fixed regardless of load, ensuring stable pressurization
- Terminal reheat coil responds to the space thermostat independently for each zone, enabling simultaneous temperature and humidity control
- ASHRAE 90.1 provides exceptions allowing reheat for airflow rates required to comply with applicable codes or accreditation standards
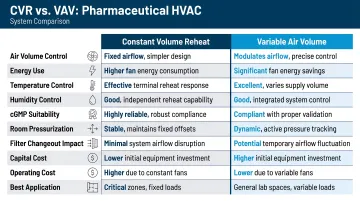
Variable Air Volume (VAV):
- Appropriate only for administrative, storage, and non-classified support areas where pressure control is not critical
- Accommodates some temperature variation without affecting product quality or compliance
- Offers meaningful energy savings compared to CVRH, making it a practical choice where regulations permit
Air Handling Unit Specifications and Location
Pharmaceutical AHUs require:
- Double-skin (sandwich) casing for a smooth, cleanable interior
- Epoxy-coated internal surfaces
- Food-grade sealants and lubricants
- Integrated drain pans with mist eliminators
- Two sets of fresh air dampers (one for 10–20% capacity, one for 100% for de-fumigation operations)
Dedicated AHUs per product suite or per hazard level are strongly recommended to prevent cross-contamination through shared ductwork.
Locate AHUs as close as possible to served spaces to minimize duct length. Place outdoor air intakes high on building sidewalls, away from exhaust stacks, truck docks, and prevailing exhaust plumes.
Validation, Qualification, and Documentation
Design Qualification (DQ)
DQ must be completed before construction begins. The DQ document:
- Identifies all HVAC systems and their functions
- Establishes critical parameters (temperature, RH, pressure differential, ACR, particle count limits) for each room
- Defines equipment performance acceptance criteria
- Includes control schematics with fail-safe positions
Per ICH Q7, DQ is the contractual and regulatory baseline against which all subsequent qualification activities are measured.
Installation Qualification (IQ) and Operational Qualification (OQ)
Installation Qualification (IQ) confirms equipment is installed per DQ specifications:
- Fan and motor data
- Filter ratings
- Calibration records
- As-built drawings
Operational Qualification (OQ) verifies system performance under designed conditions:
- Air balance testing
- HEPA filter integrity tests (DOP/PAO)
- Pressure differential verification
- Temperature mapping

According to WHO TRS 961, Annex 9, Supplement 8, temperature mapping for warehouses and ambient storage areas should run for a minimum of 7 consecutive days. For cleanrooms, 24–72 hours or longer is recommended. Calibrated data loggers should be placed throughout the space with calibration accuracy of ±0.5°C at calibration points.
Performance Qualification (PQ)
PQ validates the HVAC system under full production conditions with all equipment running and personnel present.
Ongoing GMP documentation requirements include:
- Calibration records for all sensors and controllers
- Filter replacement and integrity test logs
- Environmental monitoring reports
- Alarm history
Building Automation Systems (BAS) handle continuous environmental monitoring, automated alarming, and the audit trail FDA inspectors will review. When BAS or Environmental Monitoring Systems generate records used to demonstrate GMP compliance, 21 CFR Part 11 applies — requiring validation, audit trails, access controls, and backup copies of electronic records.
Selecting the Right A/E Partner for Pharmaceutical HVAC Design
What Differentiates a Qualified Pharmaceutical HVAC Design Partner
A qualified pharmaceutical HVAC design partner must demonstrate:
- Knowledge of FDA 21 CFR 210/211, EU GMP Annex 1, and WHO GMP requirements
- Experience writing and executing DQ/IQ/OQ/PQ protocols
- Ability to translate process requirements — heat loads, hazardous material classifications, product sensitivity — directly into HVAC system parameters
A general mechanical engineer or commercial HVAC contractor without pharmaceutical sector experience is likely to underestimate these requirements and produce a design that requires expensive post-construction modification.
These qualifications narrow the field considerably. But credentials alone don't prevent execution failures — organizational structure does.
The Case for Integrated, Multi-Disciplinary Design Teams
When HVAC is engineered separately from architecture and process design, critical coordination failures occur:
- Pressure zoning conflicts with room adjacency
- Ductwork clashes with process equipment
- BAS design is disconnected from HVAC logic
An A/E firm that integrates architecture, mechanical engineering, electrical engineering, and controls under one roof eliminates these coordination gaps and reduces overall project risk.
Hixson operates this way by design. With 75+ years of experience in pharmaceutical and Science & Technology facilities, the firm's 20 in-house technical disciplines — spanning architecture through controls and automation — work within a single project management framework built for capital-intensive regulated environments. For pharmaceutical manufacturers, that means HVAC design, process engineering, and validation support developed in coordination, not in sequence.
Frequently Asked Questions
How does HVAC system work in pharmaceutical industry?
Pharmaceutical HVAC performs four regulated functions: controlling airborne particulate and microbial contamination via HEPA filtration, maintaining differential pressure between zones to prevent cross-contamination, controlling relative humidity to protect product stability, and maintaining temperature to ensure process integrity and worker safety.
What is GMP in HVAC?
GMP (Good Manufacturing Practice) in HVAC refers to the regulatory requirements (primarily FDA 21 CFR 210/211, EU GMP Annex 1, and WHO guidelines) that govern how HVAC systems must be designed, qualified, operated, and documented in pharmaceutical manufacturing facilities. The goal is to ensure product safety and purity throughout every stage of manufacturing.
What are the 4 types of process validation?
In pharmaceutical HVAC, the four qualification/validation phases are Design Qualification (DQ), Installation Qualification (IQ), Operational Qualification (OQ), and Performance Qualification (PQ). Each phase verifies the system at a progressively deeper level, from initial design intent through full production operation.
What are the four types of HVAC systems?
The four primary HVAC system types relevant to pharmaceutical facilities are forced-air (ducted) systems, ductless mini-split systems, Variable Refrigerant Flow (VRF/VRV) systems, and heat pump systems. Pharmaceutical manufacturing predominantly uses ducted constant volume systems with terminal reheat (CVRH) for classified areas.
What are the 4 components of HVAC?
The four core HVAC components are heating source, cooling/evaporator coil, ventilation (ductwork, fans, and air distribution), and air quality control (filtration, humidification/dehumidification). Pharmaceutical applications add HEPA terminal filtration and precise control instrumentation as additional critical components.


