
Introduction
A single contamination event in a pharmaceutical or biotech facility can trigger a cascade of consequences: production shutdowns, FDA warning letters, product recalls, and the kind of regulatory scrutiny that takes years to recover from. For sterile drug manufacturers, cleanroom classification isn't an administrative checkbox — it's a direct line to product quality and patient safety.
ISO 14644 is the internationally recognized standard that governs how cleanrooms are classified, designed, tested, and monitored. Regulatory bodies worldwide — including the FDA, EU GMP, and WHO — use it as the baseline for pharmaceutical manufacturing requirements. Achieving compliance requires coordinated decisions across HVAC design, construction materials, pressure cascades, qualification protocols, and ongoing monitoring — not just an understanding of particle count limits.
This guide is written for facility engineers, quality assurance professionals, and project teams who need a practical, accurate reference. It covers:
- What ISO 14644 requires across its key parts
- How ISO classifications map to EU GMP Grades
- What compliant cleanroom design demands from the design team
- Where projects most commonly go wrong
TL;DR: Key Takeaways
- ISO 14644-1 classifies cleanrooms from ISO Class 1 (most stringent) to ISO Class 9 by maximum allowable airborne particle concentrations
- ISO 14644 and EU GMP Annex 1 are complementary but distinct: ISO governs particle classification; GMP adds microbial limits, personnel controls, and documentation
- Airflow, filtration, pressure differentials, and zone layout decisions are made early in design and are expensive to reverse
- Periodic testing alone doesn't demonstrate ongoing compliance — requalification intervals and monitoring requirements vary by class and regulatory context
- A multidisciplinary design team engaged from day one is the most reliable way to prevent compliance gaps and costly rework
What Is ISO 14644 and Why It Matters for Regulated Facilities
A Global Standard Born From Fragmentation
Before ISO 14644, cleanroom classification varied by country and industry. In the U.S., the Federal Standard 209E governed cleanroom requirements until the International Organization for Standardization (ISO) stepped in with a unified global framework. The U.S. General Services Administration officially cancelled FED-STD-209E in November 2001, with ISO 14644-1 and ISO 14644-2 designated as the replacements. Today, ISO 14644 is the reference standard across pharmaceuticals, biotechnology, medical devices, aerospace, and semiconductors.
The current governing version for classification is ISO 14644-1:2015 (Edition 2). ISO 14644-4:2022 covers design, construction, and start-up.
ISO 14644 Spans Multiple Parts — Each One Counts
Compliance requires understanding multiple parts:
- ISO 14644-1:2015 — Classification of air cleanliness by particle concentration
- ISO 14644-2:2015 — Monitoring to provide evidence of cleanroom performance
- ISO 14644-4:2022 — Design, construction, and start-up
Focusing only on Part 1 (the classification table) is a common mistake. Parts 2 and 4 carry equally important requirements for ongoing compliance and proper facility design.
Why Regulators Rely on It
Two major regulatory frameworks tie directly to ISO 14644:
- The FDA's 2004 aseptic processing guidance maps ISO 14644-1 classification to clean area requirements for sterile drug manufacturing
- EU GMP Annex 1 (revised 2022) uses ISO 14644 as the technical foundation for sterile product manufacturing standards
For facility designers and compliance teams, this alignment means ISO 14644 classification requirements must be embedded in the design program before construction begins — not reconciled during inspection.
ISO 14644 Cleanroom Classification Levels Explained
The Classification Scale
ISO 14644-1 defines nine cleanroom classes — ISO Class 1 through ISO Class 9 — where Class 1 is the most controlled and Class 9 is the least. Class is determined by maximum allowable concentrations of airborne particles per cubic meter, measured at specified particle sizes ranging from ≥0.1 µm to ≥5.0 µm.
In pharmaceutical and biotech manufacturing, ISO Classes 5, 7, and 8 are the most commonly encountered. Key particle concentration limits at ≥0.5 µm:
| ISO Class | Max Particles/m³ (≥0.5 µm) | Typical Application |
|---|---|---|
| ISO 5 | 3,520 | Aseptic filling, critical open-product zones |
| ISO 7 | 352,000 | Background for Grade A, solution prep |
| ISO 8 | 3,520,000 | Non-sterile component handling, gowning |

To put ISO 5 in context: 3,520 particles per cubic meter is roughly 100,000 times cleaner than typical indoor air.
The Three Cleanroom States
ISO 14644-1 defines three conditions under which cleanroom classification must be assessed:
- As-built — Room complete, no equipment or personnel present
- At-rest — Equipment installed and operational, no personnel
- In-operation — Normal production with personnel and processes active
This distinction has real consequences. Particle counts rise sharply when people enter a classified space — personnel are among the largest sources of contamination in cleanroom environments. GMP-regulated facilities must demonstrate compliance under in-operation conditions, not just the easier-to-achieve at-rest baseline.
Air Change Rates and HVAC Demands
ISO 14644-1 classifies particle cleanliness — it doesn't specify air changes per hour (ACH) directly. ACH requirements come from design guidance and regulatory frameworks. ISPE guidance provides typical targets by classification level:
- ISO 8: 10–20 ACH
- ISO 7: 28+ ACH in most designs
- Grade A / ISO 5: Unidirectional airflow velocity of 0.36–0.54 m/s (per EU GMP Annex 1) rather than ACH
Each step up in classification carries meaningful HVAC consequences. Decisions made early in design — duct sizing, filtration grade, supply/return layout, and redundancy — directly determine whether a facility can sustain its target class under in-operation conditions.
ISO 14644 vs. EU GMP: Understanding the Relationship
Two Frameworks, One Compliance Challenge
ISO 14644 and EU GMP Annex 1 are often conflated, but they serve different purposes:
- ISO 14644 is a technical standard for airborne particulate classification, applicable across industries
- EU GMP Annex 1 is a regulatory framework specifically for sterile medicinal product manufacturing that uses ISO classification as a baseline
ISO compliance alone does not equal GMP compliance. EU GMP Annex 1 (2022 revision) layers on microbiological limits, personnel controls, process design requirements, and documentation obligations that fall entirely outside ISO 14644's scope.
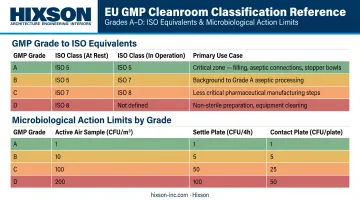
The Grade-to-ISO Mapping
| EU GMP Grade | At-Rest ISO Equivalent | In-Operation ISO Equivalent | Primary Use |
|---|---|---|---|
| Grade A | ISO 5 | ISO 5 | Aseptic filling, critical open-product exposure |
| Grade B | ISO 5 | ISO 7 | Background for Grade A operations |
| Grade C | ISO 7 | ISO 8 | Solution preparation, less critical steps |
| Grade D | ISO 8 | Not defined | Non-sterile component handling |
GMP also sets microbiological action limits not addressed by ISO at all:
| Grade | Active Air Sample | Settle Plate | Contact Plate |
|---|---|---|---|
| A | No growth | No growth | No growth |
| B | 10 CFU/m³ | 5 CFU/4h | 5 CFU/plate |
| C | 100 CFU/m³ | 50 CFU/4h | 25 CFU/plate |
| D | 200 CFU/m³ | 100 CFU/4h | 50 CFU/plate |
The In-Operation Requirement Is Non-Negotiable
GMP-regulated facilities must demonstrate compliance under real production conditions. This means monitoring systems that capture particle counts during operational cycles and correlate excursions with personnel movements, equipment activity, process steps, and gowning events. EU GMP Annex 1 requires continuous particle monitoring for Grade A zones throughout the full duration of critical processing, including equipment assembly phases.
What FDA Warning Letters Reveal
The 2024 FDA warning letter issued to Optikem International Inc. illustrates what inadequate cleanroom design and monitoring looks like in practice. Following a January 2024 inspection at the Denver facility, FDA cited 21 CFR 211.42(c)(10) for multiple failures:
- Particle board installed near HEPA filters
- Rust on filter frames and ceiling gaps compromising envelope integrity
- Monthly EM intervals insufficient for the classification level
- ISO 5 action levels that permitted up to 5 CFU
These aren't edge cases. They're the kinds of failures that happen when cleanroom design and monitoring aren't treated as integrated, ongoing commitments.
Key Design Requirements for ISO-Compliant Cleanrooms
Pressure Cascade Design
Higher-classified areas must be maintained at positive pressure relative to adjacent lower-classified zones. EU GMP Annex 1 specifies a minimum 10 Pa differential between adjacent grades, with ISPE guidance noting that 10–15 Pa is the typical FDA-recommended range when doors are closed.
Achieving this consistently requires careful engineering of:
- Airlock configurations between grade transitions
- Interlocked door systems (required for Grade A/B areas; audible/visual warning systems required for Grade C/D)
- Sealed penetrations and joints that could compromise differential pressure
- HVAC controls that maintain setpoints under dynamic conditions
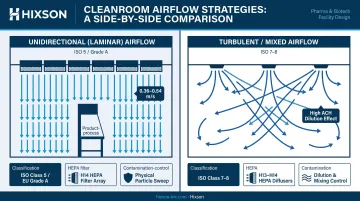
HEPA Filtration and Airflow Strategy
Two distinct airflow approaches apply at different classification levels:
- Unidirectional (laminar) airflow — Required for ISO 5/Grade A critical zones. EU GMP Annex 1 specifies 0.36–0.54 m/s at the working position. This directional flow sweeps particles away from the product/process and prevents recirculation.
- Turbulent/mixed airflow — Used in ISO 7–8 background areas, where high air change rates dilute contamination rather than directionally removing it.
HEPA filters must undergo leak testing and integrity validation at commissioning and at regular requalification intervals. ULPA filters (higher efficiency, per EN 1822-1:2019 — H14 at ≥99.995% integral efficiency, U15 at ≥99.9995%) are used in the most stringent applications.
Architectural and Material Requirements
Design decisions made during the architectural phase have compliance implications that last the life of the facility. Requirements include:
- Smooth, non-shedding, disinfectant-compatible surface finishes on walls, floors, and ceilings
- Minimal horizontal ledges where particles can accumulate
- Sealed joints and all penetrations (conduit, piping, HVAC)
- Coved corners and junctions to prevent particle trapping and facilitate cleaning
- Surface materials compatible with the cleaning and disinfection regimens the facility will use
Wall panel systems, drain configurations, and penetration details specified at schematic design are difficult and costly to correct once construction is underway.
Zone Layout and Flow Planning
Higher-classified areas belong at the facility core, surrounded by progressively lower-classified buffer zones. Material and personnel flow should move in one direction only: from lower to higher classification areas through controlled airlocks, never bypassing grade transitions.
Zone layout decisions don't exist in isolation. Pressure cascade design, HVAC strategy, and flow planning are interdependent, and coordination gaps between disciplines are a common source of compliance risk. Hixson's approach addresses this directly: with architecture, mechanical engineering, process engineering, and controls design handled by 20 in-house disciplines working from the same model simultaneously, the sequencing conflicts that arise in siloed project structures are caught before they reach construction documents.
Cleanroom Monitoring and Ongoing Qualification
Periodic Requalification vs. Continuous Monitoring
ISO 14644-2:2015 requires a monitoring plan to provide evidence of ongoing cleanroom performance. Industry practice and regulatory guidance support these requalification intervals:
- ISO Class 5 and cleaner: Every 6 months
- ISO Class 6–9: Every 12 months
But periodic classification testing alone cannot demonstrate continuous compliance between test events. EU GMP Annex 1 requires continuous particle monitoring for Grade A throughout all critical processing, and recommends routine monitoring for Grade B at sufficient frequency to detect deterioration.
What Continuous Monitoring Must Cover
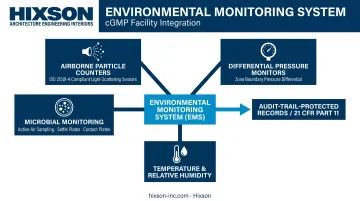
A GMP-compliant Environmental Monitoring System (EMS) — distinct from a Building Management System (BMS) — tracks and records:
- Airborne particle counts (via ISO 21501-4:2018-compliant light-scattering particle counters)
- Differential pressure across zone boundaries
- Temperature and relative humidity
- Microbial monitoring: active air sampling, settle plates, and surface contact plates (required under GMP, outside ISO 14644 scope)
Hixson's Controls & Automation team designs and integrates control systems for pharmaceutical and biotech facilities — including the data infrastructure needed to support these monitoring requirements. The team holds certifications in Rockwell Automation, AVEVA System Platform, and Ignition 8.1.
Data Integrity Requirements
EMS records must provide timestamped, audit-trail-protected data meeting requirements analogous to 21 CFR Part 11 (FDA) and EU GMP Annex 11 (EU). In practice, comprehensive automated records are what inspectors request first — and what enable defensible root cause investigations when excursions occur. Facilities that lack audit-trail-protected EMS data have faced Form 483 observations and, in serious cases, warning letters citing inadequate environmental controls.
Common Pitfalls That Compromise ISO 14644 Compliance
Design-Phase Mistakes
The most expensive cleanroom compliance failures are designed in — not introduced during operations.
- Over-classifying spaces: Specifying ISO 5 where ISO 7 suffices inflates capital cost, energy consumption, and ongoing maintenance burden with no compliance benefit
- Under-classifying spaces: Creates contamination risk and leaves the facility vulnerable during inspections
- Inadequate zone segregation: Failing to design proper pressure cascades, airlock configurations, and unidirectional flow paths from the start — these cannot be easily corrected after construction
- Late HVAC coordination: HVAC strategy that isn't integrated with architectural layout from schematic design leads to filter placement compromises and airflow dead zones
Operational Mistakes
- Assuming commissioning classification guarantees ongoing compliance — it doesn't; conditions change as processes, personnel, and equipment evolve
- Treating environmental monitoring as a documentation exercise rather than a real-time contamination control signal
- Delaying or skipping HEPA filter integrity testing and HVAC calibration between requalification cycles
- Using monthly monitoring intervals for critical zones where continuous monitoring is required (as continuous-monitoring case studies in pharma repeatedly confirm)
The Cost of Late Engagement
The decisions made during programming and schematic design — room sizing, airflow strategy, zone layout, construction materials — carry the most weight in determining compliance outcomes. Changes made during construction cost multiples of what they would have cost if addressed earlier. Changes made post-occupancy can cost an order of magnitude more.
Hixson's teams engage clients from the start, with compliance built into every phase:
- User Requirements Specification (URS) development and risk-based classification
- Design and construction documentation with HVAC, pressure cascade, and zone layout integrated from schematic design
- CQV planning with explicit IQ/OQ/PQ protocols built into design deliverables
With 75+ years designing cGMP facilities — and projects including Unither Pharmaceuticals, PETNET Solutions, and Eurofins Genomics — Hixson's engineers treat compliance as a front-end design discipline, not a final-phase checklist.
Frequently Asked Questions
What is the difference between ISO 14644 and EU GMP?
ISO 14644 is a technical standard for airborne particle classification applicable across multiple industries. EU GMP Annex 1 is a regulatory framework specifically for sterile pharmaceutical manufacturing that uses ISO classification as a baseline but adds microbiological limits, documentation requirements, and personnel controls. The two frameworks complement each other; ISO compliance alone does not satisfy GMP requirements.
What are the GMP guidelines for clean rooms?
GMP cleanroom guidelines — enforced by the FDA in the U.S. and EMA in Europe — cover all practices designed to minimize contamination risk in pharmaceutical and biotech manufacturing. Key requirements include:
- Environmental control and continuous monitoring
- Validated cleaning and disinfection procedures
- Strict gowning and personnel hygiene protocols
- Controlled material flow, comprehensive documentation, and routine audits
What are the ISO 14644 cleanroom classification levels?
ISO 14644-1 defines nine cleanroom classes (ISO 1–9) based on maximum allowable airborne particle concentrations per cubic meter at specified particle sizes. ISO Classes 5, 7, and 8 are the most commonly used in pharmaceutical and biotech manufacturing, corresponding roughly to EU GMP Grades A/B, C, and D respectively.
How often must an ISO-classified cleanroom be requalified?
Industry practice supports requalification every 6 months for ISO Class 5 and cleaner, and every 12 months for Classes 6–9. Many regulatory bodies and quality systems require at minimum annual requalification, and continuous monitoring data may support justification for adjusted intervals — but should not replace formal classification testing.
What is the difference between at-rest and in-operation cleanroom states?
At-rest means equipment is installed and operational but no personnel are present. In-operation reflects normal production conditions with personnel and processes active. Particle counts are significantly higher in-operation, and GMP-regulated facilities must demonstrate compliance under both states. Grade A zones require continuous monitoring throughout all critical processing.


