
Introduction
In pharmaceutical and biotech manufacturing, no validated process can guarantee the complete absence of viable microorganisms on every single unit — and regulators know it. "Sterile" is not a binary state; it's a probability. What manufacturers can do is reduce that probability to a scientifically defensible, regulatory-accepted level. That's exactly what Sterility Assurance Level (SAL) quantifies.
SAL sits at the center of every aseptic processing decision: facility classification, process validation, environmental monitoring frequency, and batch release criteria all flow from it. Get it wrong and you're not just facing a regulatory finding; you're risking patient safety.
This guide covers what SAL means in the context of aseptic processing and how it differs fundamentally from terminal sterilization. It also walks through the facility design, validation, and monitoring strategies that allow manufacturers to consistently hit their sterility assurance targets.
Key Takeaways
- SAL is expressed as 10⁻ⁿ — the probability of one viable organism surviving on a product unit after sterilization; 10⁻⁶ is the gold standard for terminal sterilization
- Aseptic processing achieves sterility through contamination exclusion, not terminal kill — with a PNSU typically equivalent to 10⁻³ to 10⁻⁴
- Achieving SAL targets depends on validated facility design, media fills, and environmental controls — sterility testing alone is insufficient
- Primary frameworks: FDA 21 CFR 210/211, ISO 13408, ANSI/AAMI ST67, and ISO/TS 19930
What Is Sterility Assurance Level (SAL)?
The Definition and the Math
Per ISO 11139, SAL is defined as the "probability of a single viable microorganism occurring on an item after sterilization." It's expressed in negative exponential notation: 10⁻⁶ means a 1-in-1,000,000 probability that any given unit contains a surviving viable organism after the sterilization process.
That phrasing matters. SAL doesn't describe whether a specific unit is sterile. It describes the statistical confidence you have across a population of units that have undergone a validated sterilization cycle.
As AAMI explains, absolute sterility — being "free from viable microorganisms" — cannot be proven in practice. SAL is the accepted surrogate for sterility confidence, not a guarantee.
The Counterintuitive Relationship Between Numbers and Assurance
This trips up quality professionals regularly: a lower numerical SAL represents higher sterility assurance.
- 10⁻⁶ = higher assurance than 10⁻³
- 10⁻⁷ = higher assurance than 10⁻⁶
AAMI recommends specific language to prevent confusion in regulatory discussions:
| Scenario | Recommended Language |
|---|---|
| SAL of 10⁻⁷ vs. 10⁻⁶ | "beyond" an SAL of 10⁻⁶ |
| SAL of 10⁻⁵ vs. 10⁻⁶ | "under" an SAL of 10⁻⁶ |

Avoid "higher SAL" when you mean greater sterility assurance — it creates the exact ambiguity you want to eliminate.
SAL vs. PNSU: Two Different Tools
SAL is a mathematical value derived from measurable log-reduction of a known bioburden. It applies strictly to terminal sterilization processes where microbial populations are actively and quantifiably reduced.
PNSU (Probability of a Nonsterile Unit) is the corresponding concept for aseptic processing, where sterility is maintained through contamination exclusion rather than active microbial reduction. Both are expressed as 10⁻ⁿ, but they are not interchangeable: ISO 11139 and ANSI/AAMI ST67 formalize this distinction explicitly.
How SAL Applies Differently to Aseptic Processing vs. Terminal Sterilization
Terminal Sterilization: Active Log-Reduction
In terminal sterilization, the product is filled and sealed in its final container, then subjected to a validated sterilization cycle — moist heat, radiation, ethylene oxide, or vaporized hydrogen peroxide. The cycle logarithmically reduces the starting bioburden to achieve a measurable 10⁻⁶ SAL.
Because the reduction is active and reproducible, it can be mathematically validated. ISO 13408-1 and FDA guidance both express a clear preference for terminal sterilization precisely because of this measurability.
Aseptic Processing: Contamination Exclusion
Aseptic processing takes a fundamentally different approach. The drug product, container, and closure are each sterilized separately by appropriate methods — sterilizing filtration for liquids, dry heat or moist heat for containers and closures — then assembled in a controlled environment. No sterilization step follows assembly — sterility depends entirely on keeping contamination out, not eliminating it after the fact.
That distinction has a direct SAL consequence: aseptic processing cannot produce a measurable 10⁻⁶ SAL because contamination risk during assembly cannot be reduced to zero. Industry and regulatory consensus places the achievable PNSU at 10⁻³ to 10⁻⁴.
ISO/TS 19930 presents a flowchart positioning aseptic processing and terminal sterilization with alternative SALs as equivalent options when 10⁻⁶ terminal sterilization cannot be achieved — a direct acknowledgment that the two pathways serve the same patient safety goal through different means.
When Aseptic Processing Is the Right Choice
That equivalence matters most when terminal sterilization would damage the product itself. Regulatory agencies accept aseptic processing as appropriate in these situations:
- Heat-sensitive biologics such as monoclonal antibodies, vaccines, and recombinant proteins
- Combination products where the device or delivery system cannot withstand sterilization conditions
- Parenterals where moist heat or radiation would degrade the active ingredient
- Cell-based therapies subject to distinct regulatory frameworks
Alternative SALs: A Third Pathway
ANSI/AAMI ST67:2019 and ISO/TS 19930 also recognize alternative SALs of 10⁻³, 10⁻⁴, and 10⁻⁵ for terminal sterilization of products that cannot withstand full 10⁻⁶ TS conditions. Research by Srun et al. published in PubMed found no statistically significant difference in surgical site infection incidence across SALs of 10⁻⁴ to 10⁻⁶ — clinical data that underpins the regulatory acceptance of these alternative pathways.
Cleanroom Classification and Facility Design: The Engineering Foundation of SAL
ISO/FDA Air Classification Zones
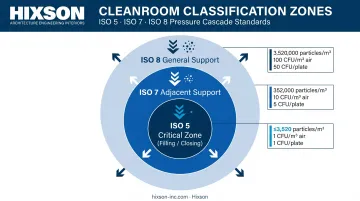
The physical environment is the first line of defense in aseptic processing. The FDA's 2004 Aseptic Processing Guidance establishes three classification zones, each with defined particle and microbiological limits:
| Zone | ISO Class | ≥0.5 µm Particles/m³ | Active Air Limit | Contact Plate Limit |
|---|---|---|---|---|
| Critical (filling/closing) | ISO 5 | 3,520 | 1 CFU/m³ | 1 CFU/plate |
| Adjacent support | ISO 7 | 352,000 | 10 CFU/m³ | 5 CFU/plate |
| General support | ISO 8 | 3,520,000 | 100 CFU/m³ | 50 CFU/plate |
The ISO 5 critical zone — where sterile product, containers, and closures are exposed — should normally yield no microbiological contaminants during sampling. Meeting those limits depends on three engineering systems working in concert.
Airflow, Pressure, and HEPA Requirements
Unidirectional Airflow (UDAF)
- HEPA-filtered air at approximately 0.45 m/s (±20%) sweeps particles away from exposed product
- In situ smoke studies under dynamic conditions are required to confirm unidirectional flow and absence of turbulence or eddy currents
- Turbulence near the fill point is a direct contamination pathway — smoke studies, not calculations, prove absence
Pressure Cascade Design
- Minimum 10–15 Pa positive pressure differential between adjacent rooms of differing classification
- Pressure flows from cleanest to least clean zones — any reversal creates an inward contamination pathway
- Where an unclassified room is adjacent to an aseptic area, FDA recommends at least 12.5 Pa overpressure
- Continuous monitoring with rapid alarm response is required; pressure deviations during filling are critical events
HEPA Filter Integrity
- Leak testing at installation and at least semi-annually for aseptic processing rooms
- Additional testing required following any facility disturbance
- Any integrity failure — regardless of how minor — requires filter replacement before resuming production
Achieving these environmental targets requires that HVAC design, cleanroom zoning, and pressure cascade work as a single coordinated system — not as sequential handoffs between disciplines.
Hixson's multi-discipline teams work in parallel from initial layout through commissioning and qualification (CQV), so that architecture, mechanical engineering, process engineering, and controls are aligned from the start rather than reconciled at the end.
Validating the Aseptic Process: Media Fills, Filtration, and Sterilization
Media Fills: The Primary Validation Tool
A media fill (process simulation) replaces the drug product with soybean casein digest medium (SCDM), a microbiological growth medium that supports a broad range of bacteria, yeast, and mold. Units are filled under normal production conditions, sealed, and incubated at 20–35°C for a minimum of 14 days.
The simulation must reflect worst-case operations: representative interventions, actual line speeds, shift changes, and all personnel who are authorized to enter the aseptic area.
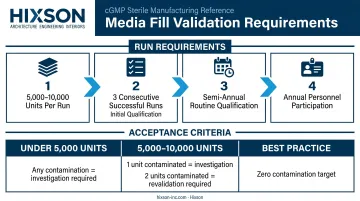
Run sizing and frequency requirements (per FDA):
- Acceptable starting point: 5,000–10,000 units per run
- Initial qualification: at least three consecutive successful runs
- Routine qualification: semi-annually per processing line
- All authorized personnel must participate at least annually
Acceptance criteria:
- Fewer than 5,000 units: any contaminated unit triggers investigation and requires consideration of revalidation
- 5,000–10,000 units: one contaminated unit requires investigation; two units = revalidation
- Modern aseptic operations are expected to approach zero contamination; the FDA considers any failure objectionable regardless of statistical thresholds
Media fill results feed directly into the broader validation picture — but filtration and equipment sterilization require their own documented evidence.
Sterilizing Filtration Validation
For heat-sensitive liquid drug formulations, sterilizing filtration is the primary sterilization method. Key parameters:
- 0.2 µm rated filters, validated with a minimum challenge of 10⁷ organisms/cm² of effective filtration area
- Challenge organism: Brevundimonas diminuta (ATCC 19146), per ASTM F838
- Forward flow and bubble point integrity tests performed post-use (pre-use/post-sterilization testing also recommended by FDA where feasible)
- Any post-use filter failure may indicate a sterility breach and triggers investigation and batch disposition review
Sterilization Validation for Equipment, Containers, and Closures
Every surface that contacts sterile product must be validated:
- Heat distribution and penetration studies with calibrated sensors and biological indicators at the most difficult-to-sterilize locations
- FDA requires a demonstrated SAL of 10⁻⁶ or better for all sterilized equipment surfaces
- Revalidation triggers include: equipment changes, facility modifications, adverse environmental trends, or sterility test failures
Environmental Monitoring and Regulatory Compliance for Ongoing Sterility Assurance
Environmental Monitoring as a Control System
An environmental monitoring (EM) program functions as an active control system, not a documentation exercise. FDA requires EM programs to cover air, surfaces, floors, walls, equipment, and personnel (including gloves and gowns).
Effective EM programs share several characteristics:
- Sample air with impaction or centrifugal devices during each production shift
- Sample surfaces and personnel in critical and adjacent zones
- Set distinct alert and action levels that separate investigation-worthy trends from events requiring immediate response
- Track data over time — single data points are rarely meaningful; trending reveals whether contamination controls are drifting
- Investigate promptly and execute CAPA when action levels are exceeded or adverse trends emerge
The Regulatory Framework
The primary documents governing SAL and aseptic processing in the US:
| Standard | Scope |
|---|---|
| FDA 2004 Guidance (21 CFR 210/211) | Sterile drug products via aseptic processing; reflects current CGMP expectations |
| ISO 13408 (Parts 1–8) | Aseptic processing of health care products across all modalities |
| ISO 14644-1 | Cleanroom air cleanliness classification |
| ANSI/AAMI ST67:2019 | SAL selection guidance for products labeled "sterile" |
| ISO/TS 19930 | Alternative SAL pathways for products unable to achieve 10⁻⁶ TS |
FDA guidance is non-binding but describes current agency expectations — deviating from it requires documented scientific justification.
Why Sterility Testing Alone Is Not Sufficient
USP <71> sterility testing is a batch release tool, not a sterility assurance system. The statistical limitation is stark: according to FDA's own guidance, a 10,000-unit lot with a 0.1% contamination rate has approximately a 98% chance of passing sterility testing using a standard 20-unit sample.
That means a contaminated batch can — and statistically will — pass sterility testing most of the time.
The true sterility assurance system is the validated aseptic process, the environmental monitoring program, and the controlled facility design working in combination. Sterility testing is a release checkpoint — the process controls are what actually prevent contamination.

Frequently Asked Questions
What is the sterility assurance level for aseptic processing?
Aseptic processing does not produce a formally measured SAL. Instead, it achieves a Probability of a Nonsterile Unit (PNSU) generally equivalent to 10⁻³ to 10⁻⁴, maintained through validated contamination-exclusion controls — separate sterilization of components, controlled filling environments, and ongoing environmental monitoring — not through active microbial reduction.
Why is 3% SCDM used in media fill?
Soybean casein digest medium at approximately 3% concentration (30 g/L total solids per USP <71>) supports growth of gram-positive and gram-negative bacteria, yeast, and mold. This broad-spectrum coverage makes it capable of detecting contamination from the wide range of microorganisms that could be introduced during aseptic assembly.
What is the difference between SAL and PNSU in aseptic processing?
SAL applies to terminal sterilization, where microbial populations are logarithmically reduced from a known starting bioburden. PNSU applies to aseptic processing, where sterility is maintained by exclusion rather than elimination. Both are expressed as 10⁻ⁿ but represent fundamentally different mechanisms and are not interchangeable.
What cleanroom classification is required for aseptic filling operations?
The critical zone where sterile product, containers, and closures are exposed must meet ISO 5 (Class 100) standards. The immediately adjacent area requires at minimum ISO 7 (Class 10,000). Supporting areas operate at ISO 8 (Class 100,000) per FDA guidance and ISO 14644-1.
How often should media fills be performed for aseptic processing lines?
FDA requires at least three consecutive successful runs during initial line qualification and semi-annual runs thereafter. Additional media fills are required following significant process changes, facility modifications, or any sterility failure investigation.
When is aseptic processing preferred over terminal sterilization?
Aseptic processing is used when a product cannot withstand terminal sterilization without loss of efficacy or safety. This includes heat-sensitive biologics, live-cell products, and combination products where moist heat, radiation, or chemical sterilization would degrade the active ingredient or compromise device integrity.


