
Introduction
APIs are the biologically active core of every medicine. A purity deviation of even a fraction of a percent can render a drug ineffective — or dangerous. That's not theoretical: the 2007–2008 heparin contamination crisis and the 2018 valsartan nitrosamine scandal both traced back to manufacturing failures at the API level, triggering mass recalls, drug shortages, and patient harm across multiple countries.
ICH Q7 exists because finished drug product GMP standards weren't enough. Published in 2000 by the International Council for Harmonisation, Q7 establishes the GMP framework specifically for API manufacturing — and it has since been adopted by the FDA, EMA, WHO, Health Canada, and regulatory bodies worldwide.
That adoption shapes real facility decisions — which is what this guide addresses. It covers Q7's scope, key section requirements, facility design implications, and the most common compliance gaps. It's written for:
- Quality managers and compliance leads
- Manufacturing and process engineers
- Facility designers working in pharma and biotech API environments
Key Takeaways: ICH Q7 At a Glance
- ICH Q7 is the global GMP standard for APIs, adopted by the FDA (as Q7A), EMA (as EudraLex Part II), WHO, and major regulators worldwide
- It governs the full API lifecycle — from designated starting materials through production, testing, packaging, storage, and distribution
- Core requirements: independent Quality Unit, validated processes, strict documentation, supplier qualification, and annual product reviews
- Applies to API manufacturers, contract manufacturers, testing labs, and companies sourcing APIs globally
- Non-compliance consequences include FDA warning letters, import bans, recalls, and drug shortages
What Is ICH Q7 and Why It Matters
ICH Q7 is a guideline issued by the International Council for Harmonisation in November 2000 that establishes Good Manufacturing Practice requirements specifically for APIs. Before Q7, GMP standards focused primarily on finished drug products — leaving a significant gap in regulatory coverage for the active ingredients themselves.
Q7's core objective: ensure that APIs consistently meet the quality and purity characteristics they are purported to possess, under a documented and auditable quality management system.
Technically a Guideline, Practically Mandatory
While Q7 is technically a guideline rather than a binding regulation, its requirements are embedded in national law across major markets:
- United States: FDA 21 CFR framework, adopted as guidance Q7A (2001)
- European Union: EudraLex Volume 4 Part II
- Global: WHO Technical Report Series 957 Annex 2
Any company selling APIs in these markets must comply. Non-compliance invites import alerts, warning letters, and market exclusion.
The Stakes Are Real
The global API market was estimated at $255 billion in 2024, projected to reach $359 billion by 2030. At that scale, manufacturing failures don't stay contained.
The 2007–2008 heparin crisis, traced to oversulfated chondroitin sulfate contamination in Chinese-sourced starting materials, was linked to 81 reported deaths and hundreds of adverse reactions. FDA findings cited inadequate GMP controls at the API manufacturer — a textbook Q7 gap.
A decade later, the 2018 valsartan contamination involved NDMA (a probable human carcinogen) in APIs from Zhejiang Huahai Pharmaceuticals. The FDA's warning letter cited failure to evaluate process changes for mutagenic impurity formation. Approximately 2,300 batches across 24 countries were recalled.
Scope: What ICH Q7 Covers — and What It Doesn't
What Q7 Covers
Per the ICH Q7 guideline, the standard applies to APIs manufactured by:
- Chemical synthesis
- Extraction from natural sources
- Fermentation and cell culture
- Recovery from natural sources
- Hybrid combinations of the above
It also applies to sterile APIs — up to the point immediately before the API is rendered sterile.
Where Q7 Requirements Begin
Q7 applies from the designated API starting material onward. Compliance intensity increases at each successive step closer to the final API. Steps prior to the introduction of starting materials are expected to have "appropriate controls," but aren't subject to full Q7 GMP requirements.
Where that starting material designation is placed is one of the most common compliance disputes — covered in the compliance challenges section below.
What Q7 Explicitly Excludes
Q7 does not apply to:
- Finished drug product manufacturing (covered by 21 CFR Parts 210–211 or EudraLex Part I)
- Vaccines, whole blood, plasma derivatives, and gene therapy APIs
- Radiopharmaceuticals
- Medical gases
- Bulk-packaged drug products
Key Requirements of ICH Q7: A Section-by-Section Overview
ICH Q7 spans 20 sections. Here's what each major area demands.
Quality Management System and Quality Unit
Q7 requires an independent Quality Unit with documented authority to:
- Approve or reject raw materials, intermediates, and final APIs
- Conduct deviation investigations
- Oversee batch release
The Product Quality Review requirement mandates an annual review of all batches, deviations, out-of-specification (OOS) results, and changes for each API — designed to detect trends and drive continuous improvement.
Personnel, Training, and Hygiene
Q7 requires sufficient qualified personnel with clearly defined roles. Specific requirements include:
- Documented GMP training programs, updated when procedures or equipment change
- Appropriate gowning and hygiene protocols
- Restricted access to manufacturing areas
- Exclusion of personnel who could compromise product quality
Materials Management and Supplier Qualification
Q7 formalizes the concept of "API starting materials" — the designated raw materials or intermediates that enter the controlled manufacturing process. Requirements include:
- Written specifications for all incoming materials
- Quarantine procedures pending testing
- Formal supplier qualification, including audits and Certificate of Analysis (CoA) verification
The heparin crisis is the textbook example of what happens when these controls fail. FDA findings confirmed that the Chinese manufacturer lacked adequate impurity-removal processes, unclean equipment, and inadequate systems to ensure raw-ingredient quality from outside suppliers.
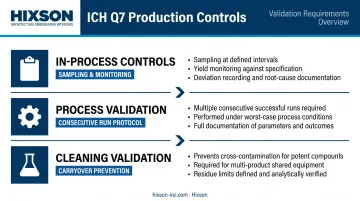
Production, In-Process Controls, and Validation
Every manufacturing step must be executed against detailed written instructions (batch/master records). Validation requirements extend to:
- Documented in-process sampling, yield monitoring, and deviation recording
- Process validation across multiple consecutive successful production runs under worst-case conditions
- Cleaning validation — particularly for potent or multi-product equipment — to prevent carryover between batches
Laboratory Controls and Data Integrity
Q7's QC laboratory requirements cover:
- Validated analytical test methods
- Traceable reference standards
- Stability testing programs with defined retest/expiry dates
- Sample retention
Regulators have increasingly focused on ALCOA+ principles — Attributable, Legible, Contemporaneous, Original, Accurate, plus Complete, Consistent, Enduring, and Available. All analytical records must include full audit trails, and electronic systems must be validated to prevent data manipulation.
Recent FDA warning letters show what non-compliance looks like in practice. A 2024 warning letter to Sichuan Deebio Pharmaceutical cited QC personnel relying on memory for test results and non-contemporaneous laboratory documentation. A 2020 letter to International Trading Pharm Lab cited the ability to delete raw data files and alter timestamps — a fundamental ALCOA+ failure.
Change Control, Deviations, and Continuous Improvement
Q7 requires a formal change management system: all proposed changes to processes, equipment, materials, or methods must be reviewed by the Quality Unit, documented, and evaluated for quality impact before implementation. This connects directly to the valsartan crisis — Zhejiang Huahai failed to evaluate how a process change affected mutagenic impurity formation.
CAPA (Corrective and Preventive Action) systems must address root causes, not just symptoms. Where Q7 sets the baseline, ICH Q10 — the Pharmaceutical Quality System guideline — provides the broader framework for sustaining that discipline: building quality culture, management review, and knowledge management into ongoing operations.
Facility and Equipment Design Requirements Under ICH Q7
Q7's facility and equipment requirements aren't abstract policy — they translate directly into architectural and engineering decisions that must be made before a facility is built. Getting them wrong means expensive retrofits and failed inspections.
Facility Layout and Material Flow
Manufacturing areas must be designed to enable logical, one-way flow of materials and personnel, from raw materials through production to packaging, to prevent cross-contamination and mix-ups. Separate, dedicated areas are required for:
- Weighing and dispensing
- Reaction and synthesis
- Packaging and labeling
- In-process and finished goods storage
- Quality control sampling

Airlocks, pressure differentials, and access controls are essential design elements determined during architectural and process layout planning, not during construction.
HVAC, Utilities, and Critical Systems
Q7 expects validated critical utilities including:
- Water systems: Purified water, Water for Injection (WFI) where applicable
- HVAC: Appropriate filtration, pressure cascade controls, and environmental monitoring
- Compressed air and clean steam
These systems must be monitored and documented: requirements that must be built into the facility's mechanical and plumbing systems from Day 1.
Hixson's pharma and biotech work covers the full utility stack: WFI, USP purified water, clean steam, pure gases, and HVAC systems with pressurization and filtration appropriate to the classified space. Design documentation is structured for validation-readiness from the URS phase, so qualification activities don't require reverse-engineering design intent.
Equipment Qualification
All critical equipment must undergo Installation Qualification (IQ), Operational Qualification (OQ), and Performance Qualification (PQ) before commercial use. Equipment design choices that directly affect how straightforwardly a facility achieves these qualifications include:
- Material of construction (surface finish, corrosion resistance)
- Cleanability (CIP-ability, dead-leg elimination)
- Containment features
- Instrument calibration infrastructure
Hixson's process engineering and controls teams produce P&IDs, equipment specifications, and URS-to-IQ traceability documentation — giving qualification teams a direct line from design intent to acceptance criteria.
Potent API Containment
For highly potent or sensitizing compounds, Q7 requires enhanced containment, segregated suites, and dedicated equipment. Containment strategies progress through a hierarchy:
- Open handling (appropriate for lower-potency compounds with administrative controls)
- Closed systems (gloveboxes, closed transfers)
- Isolators (for high-potency APIs requiring the highest level of operator protection)
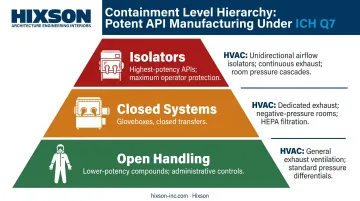
Each tier carries HVAC and engineering control requirements that must be incorporated into facility design. Hixson has designed containment solutions across this spectrum — isolators, biosafety cabinets, and BSL-2 suites — each with dedicated HVAC and waste management systems sized to the compound's occupational exposure band.
Common Compliance Challenges and How to Avoid Them
Documentation and Data Integrity Failures
Poor documentation remains one of the most cited reasons for FDA 483 observations at API facilities. Common failures include:
- Missing or incomplete audit trails
- Batch records with gaps or corrections that lack signatures and dates
- Electronic data that can be overwritten or deleted without a record
- Paper records completed after the fact
How to address it: Implement electronic batch records and quality management systems built around ALCOA+ principles. Establish SOPs that make compliant documentation the default — not the exception — for operators. Ensure computerized systems undergo validation that prevents data manipulation.
Inadequate Supplier and Starting Material Controls
Many API quality failures originate upstream. Relying solely on supplier-provided CoAs without independent verification compounds that risk.
How to address it: Build a risk-based supplier qualification program that includes:
- Facility audits (on-site or remote)
- Full specification testing of incoming materials, not just CoA acceptance
- Ongoing supplier performance monitoring
- Heightened scrutiny for starting materials that represent significant structural fragments of the final API molecule
A 2023 FDA warning letter to ALI Pharmaceutical Manufacturing cited API lots released based on CoAs marked "not suitable for GMP release" — a direct failure of this requirement.
Starting Material Definition Disputes
One of the most consistent compliance ambiguities under Q7 is where in the synthesis the API starting material designation falls. Regulators — particularly the FDA and EMA — scrutinize whether companies have pushed this designation too late in the synthesis to reduce the scope of GMP activities.
Work with quality and regulatory teams early in process development to establish a scientifically justified starting material designation. A defensible designation is one where the material represents a significant structural fragment, has established specifications, and is commercially available from qualified suppliers.
This conversation should happen during process development — not during a pre-approval inspection.
ICH Q7 in a Global Regulatory Context
ICH Q7 has been adopted across every major regulatory region, making it the effective global baseline for API GMP:
| Region | Implementation |
|---|---|
| United States | FDA Q7A guidance (2001) + Q&A guidance (2018) |
| European Union | EudraLex Volume 4 Part II |
| Global/Developing | WHO TRS 957 Annex 2 (2010) |
| Canada | Health Canada adoption |
| Japan | PMDA implementation |

That shared baseline also means compliance failures carry cross-border consequences. Enforcement activity has intensified across all major regions in recent years.
Current Enforcement Trends
Inspection frequency and scrutiny have increased across all major regions:
- Unannounced foreign inspections: FDA has expanded its use of for-cause and unannounced inspections at overseas API manufacturing sites
- EU-US Mutual Recognition Agreement: Fully operational for human medicines since July 2019, cross-recognition of inspection results means a finding by one authority can have consequences in both markets
- FDA Quality Management Maturity (QMM) program: A voluntary program encouraging manufacturers to go beyond minimum cGMP compliance — cGMP is a prerequisite, not the ceiling
- Drug shortage linkage: The FDA Drug Shortages Task Force explicitly links underinvestment in manufacturing quality to supply disruptions — connecting Q7 compliance failures to real-world patient access consequences
For API manufacturers operating across multiple markets, the practical takeaway is clear: a single inspection finding can now affect market access in the US, EU, and beyond simultaneously.
Frequently Asked Questions
What is the ICH Q7 good manufacturing guideline?
ICH Q7 is an internationally harmonized GMP guideline for API manufacturing, issued by the International Council for Harmonisation in November 2000. It has been adopted by the FDA, EMA, WHO, and other major regulators to ensure APIs consistently meet quality and purity standards throughout production.
What are the Q7 standards?
Q7 covers 20 sections spanning quality management systems, personnel and training, facility and equipment design, materials management, production controls, laboratory controls, validation, documentation, change control, and packaging. Controls become progressively stricter as manufacturing steps approach the final API.
What does Q7 guidance not apply to?
Q7 does not apply to finished drug product manufacturing, radiopharmaceuticals, vaccines, whole blood and plasma derivatives, gene therapy APIs, or steps occurring before the designated API starting material is introduced. Appropriate quality controls are still expected for those pre-starting material steps.
What is the FDA guidance Q7?
The FDA adopted ICH Q7 as guidance for industry under the designation "Q7A" in August 2001, then issued a companion Questions and Answers guidance in April 2018. Both documents are enforceable through the agency's cGMP inspection program.
What are the 5 pillars of GMP?
The most commonly cited pillars are: people (qualified and trained personnel), premises (facilities and equipment fit for purpose), processes (validated and consistently followed procedures), products (defined quality specifications), and documentation (accurate and complete records). All five are directly addressed within ICH Q7 for API manufacturing.


